Article body
Le glucose est un des substrats énergétiques obligatoires d’un certain nombre de tissus, comme les hématies, la medulla rénale et le cerveau. Ce dernier utilise chez l’homme environ 120 g de glucose par jour. Un apport continu de glucose est donc une condition absolue de notre survie et l’organisme a développé des stratégies lui permettant de faire face au caractère discontinu des apports nutritionnels. Après le repas, le glucose arrivant en abondance est mis en réserve sous forme de glycogène dans les organes, en particulier dans le foie. Dans le foie et les muscles, ce processus est contrôlé par l’insuline, sécrétée en cas d’absorption glucidique. À distance des repas, le foie libère du glucose à partir du glycogène (glycogénolyse) puis si la période de jeûne se prolonge (quelques heures), le foie met en route une synthèse de novo de glucose appelée néoglucogenèse permettant de fabriquer du glucose à partir des acides aminés contenus dans les protéines (cela permet de comprendre pourquoi le jeûne s’accompagne d’une fonte musculaire). L’insuline sécrétée au moment du repas inhibe la glycogénolyse et la gluconéogenèse, évitant ainsi un apport simultané endogène et exogène de glucose et l’hyperglycémie qui pourrait en résulter. L’insuline a donc un rôle majeur dans le maintien de l’homéostasie glucidique par ses actions directes sur le foie.
Le mécanisme de sécrétion de cette hormone lorsque la glycémie s’élève fait schématiquement intervenir une augmentation de l’utilisation de glucose par la cellule β-pancréatique, une production accrue d’ATP et une diminution du rapport ADP/ATP conduisant à la fermeture de canaux K+ ATP-dépendants (Figure 1). Cela entraîne une dépolarisation cellulaire qui permet l’ouverture de canaux Ca2+ dépendant du voltage. L’augmentation du calcium intracellulaire, de concert avec d’autres seconds messagers (AMPc), stimule la libération d’insuline.
Figure 1
Le canal K+ pancréatique dépendant de l’ATP.
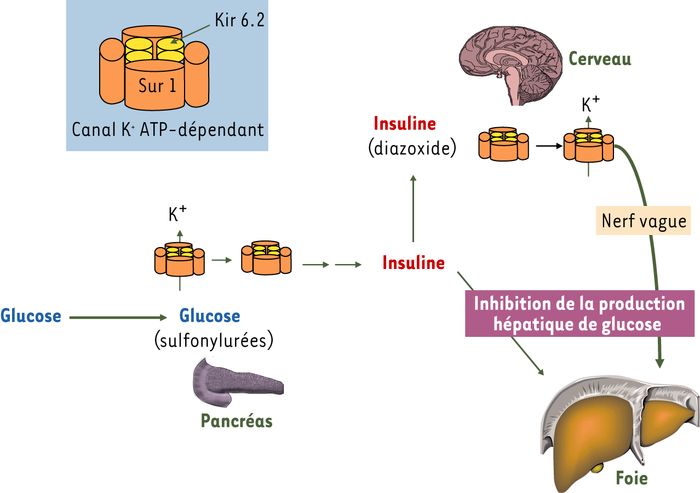
Le glucose absorbé au moment du repas est utilisé par la cellule β-pancréatique, ce qui conduit à la fermeture des canaux potassiques (voir les sous-unités du canal dans l’encart) et à la sécrétion d’insuline. L’effet du glucose, la fermeture des canaux K+, peut être mimé par les sulfonylurées. L’insuline inhibe, par des effets directs sur la cellule hépatique, la production de glucose. L’insuline agit aussi sur le cerveau au niveau de l’hypothalamus en entraînant dans une population de neurones l’ouverture des canaux K+. Cela déclenche un signal, relayé par le nerf vague, qui entraîne également l’inhibition de la production hépatique de glucose. L’effet de l’insuline sur l’ouverture des canaux potassiques peut être mimé par le diazoxide.
Le canal K+ pancréatique dépendant de l’ATP (Figure 1) est un hétéro-octamère formé de quatre sous-unités appelés Kir 6.2 (inwardly rectifying K+ channel), le canal ionique proprement dit et de quatre sous-unités régulatrices appelées SUR 1 (sulfonylurea receptor), de la famille des transporteurs ABC (ATP-binding cassette) [1]. C’est en se liant à ces sous-unités que les drogues de la famille des sulfonylurées (par exemple le tolbutamide ou le glibenclamide) utilisées dans le traitement du diabète de type 2 ferment le canal potassique et stimulent la sécrétion d’insuline. Inversement, le diazoxide en se liant à SUR 1 ouvre le canal potassique et inhibe la sécrétion d’insuline. Le même type de canal est également présent dans les neurones alors que des isoformes différentes de SUR (SUR 2A et B) sont présentes dans les muscles squelettiques, le muscle cardiaque et les muscles lisses [2]. Les isoformes SUR 2 ont une affinité beaucoup plus faible pour les sulfonylurées que SUR 1.
On sait, depuis Claude Bernard et sa célèbre « piqûre » du plancher du 4e ventricule entraînant un diabète transitoire, que le cerveau peut contrôler l’homéostasie glucidique. Bien que l’utilisation du glucose dans le cerveau ne soit pas dépendante de l’insuline (ce qui entraînerait un fonctionnement cérébral assez chaotique puisqu’il dépendrait de l’absorption de glucides !), il a été montré que l’insuline pouvait avoir une action centrale sur le métabolisme énergétique, en diminuant la prise alimentaire et en favorisant la dépense énergétique [3]. L’insuline peut également exercer au niveau central une action sur le métabolisme glucidique périphérique et en particulier hépatique. En effet, une injection intra-cérébroventriculaire d’insuline diminue la production hépatique de glucose [4], alors que le blocage de la signalisation insulinique au niveau hypothalamique a l’effet inverse. Rappelons que l’hypothalamus contient, au sein de noyaux spécifiques, des neurones exerçant un effet anabolique (stimulation de la prise de nourriture, diminution de la dépense énergétique), ou catabolique (augmentation de la dépense énergétique, inhibition de la prise alimentaire). En 2000, T.S. Zheng et al. ont montré que l’insuline pouvait diminuer par hyperpolarisation l’activité d’une sous-population de neurones dans les noyaux arqués et ventromédians de l’hypothalamus, en ouvrant des canaux K+ dépendants de l’ATP [5]. Cette observation vient d’être prolongée en montrant que la modulation de l’activité de ces canaux entraînait des modifications très importantes du métabolisme glucidique hépatique chez le rat. Pocai et al. [6] ont utilisé un activateur du canal, le diazoxide délivré par voie intracérébroventriculaire ou par voie intrahypothalamique. Cela entraîne une hypoglycémie liée à une inhibition de la production hépatique de glucose et en particulier de la gluconéogenèse, un effet très semblable à celui de l’insuline administrée au niveau hypothalamique. Les effets inhibiteurs centraux de l’insuline sur la production hépatique de glucose peuvent être supprimés en utilisant une sulfonylurée bloquant le canal K+ ou chez des souris dont le gène SUR 1 a été inactivé. Les auteurs ont enfin démontré que les branches efférentes hépatiques du nerf vague étaient nécessaires à la transmission du signal insulinique du cerveau vers le foie. Le mécanisme de signalisation qui va du récepteur de l’insuline au canal potassique reste mal connu. Il fait probablement intervenir IRS 2, une protéine qui se lie au récepteur de l’insuline activé par la liaison de l’hormone [7]. IRS 2 est ensuite phosphorylé sur des résidus tyrosines par l’activité tyrosine kinase du récepteur et recrute alors des effecteurs intracellulaires. Un de ces effecteurs, la phosphatidylinositol-3-kinase, est également impliqué dans les effets hypothalamiques de l’insuline [6]. Toutefois, les étapes ultérieures restent inconnues.
En résumé, l’élévation de glucose au moment des repas entraîne la sécrétion d’insuline qui outre ses effets directs sur le métabolisme hépatique active un circuit neuronal central inhibant la production hépatique de glucose (Figure 1).
Les canaux K+ dépendants de l’ATP sont donc impliqués dans le système de régulation par l’insuline du métabolisme glucidique au niveau de la sécrétion de l’hormone mais également au niveau de son action hypoglycémiante.
Plus de cinquante mutations dans l’une ou l’autre sous-unité du canal K+ dépendant de l’ATP ont été décrites chez l’homme [1]. Elles sont responsables d’une forme récessive d’hyper-insulinisme persistant de l’enfant qui se caractérise par le découplage de l’actvité électrique de la cellule β-pancréatique et du métabolisme glucidique. On peut se demander si ces mutations ont également des conséquences sur la régulation centrale de la production hépatique de glucose et sur la sensibilité à l’insuline [8].
Il est bien sûr tentant au vu de ces informations d’utiliser une molécule « ouvrant » les canaux K+ dépendants de l’ATP (comme le diazoxide) pour diminuer la production hépatique de glucose, un des principaux responsables de l’hyperglycémie observée lors du diabète. Il faut toutefois se rappeler que ces mêmes canaux K+ doivent être fermés dans les cellules β du pancréas pour permettre la sécrétion d’insuline en réponse au glucose. Les diabétiques de type 1 sans insulinosécrétion résiduelle pourraient cependant représenter une population de patients chez laquelle on pourrait envisager d’utiliser un tel traitement pour diminuer la production hépatique de glucose sans risquer évidemment de détérioration de la sécrétion. On peut à l’inverse se demander si le bénéfice bien établi en terme de sécrétion d’insuline d’un traitement du diabète de type 2 par les sulfonylurées ne pourrait être en fait amoindri par une action anti-insulinique centrale.
Appendices
Références
- 1. Aguilar-Bryan L, Bryan J, Nakazaki M. Of mice and men: K(ATP) channels and insulin secretion. Recent Prog Horm Res 2001 ; 56 : 47-68.
- 2. Aguilar-Bryan L, Bryan J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocrinol Rev 1999 ; 20 : 101-35.
- 3. Woods SC, Lotter EC, McKay LD, Porte D Jr. Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature 1979 ; 282 : 503-5.
- 4. Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med 2002 ; 8 : 1376-82.
- 5. Spanswick D, Smith MA, Mirshamsi S, et al. Insulin activates ATP-sensitive K+ channels in hypothalamic neurons of lean, but not obese rats. Nat Neurosci 2000 ; 3 : 757-8.
- 6. Pocai A, Lam TK, Gutierrez-Juarez R, et al. Hypothalamic K(ATP) channels control hepatic glucose production. Nature 2005 : 434 :1026-31.
- 7. Choudhury AI, Heffron H, Smith MA, et al. The role of insulin receptor substrate 2 in hypothalamic and beta cell function. J Clin Invest 2005 : 115 : 940-50.
- 8. Gribble FM. Metabolism: a higher power for insulin. Nature 2005 ; 434 : 965-6.
List of figures
Figure 1
Le canal K+ pancréatique dépendant de l’ATP.
Le glucose absorbé au moment du repas est utilisé par la cellule β-pancréatique, ce qui conduit à la fermeture des canaux potassiques (voir les sous-unités du canal dans l’encart) et à la sécrétion d’insuline. L’effet du glucose, la fermeture des canaux K+, peut être mimé par les sulfonylurées. L’insuline inhibe, par des effets directs sur la cellule hépatique, la production de glucose. L’insuline agit aussi sur le cerveau au niveau de l’hypothalamus en entraînant dans une population de neurones l’ouverture des canaux K+. Cela déclenche un signal, relayé par le nerf vague, qui entraîne également l’inhibition de la production hépatique de glucose. L’effet de l’insuline sur l’ouverture des canaux potassiques peut être mimé par le diazoxide.