Article body
Chez les eucaryotes, la source principale de formes réactives de l'oxygène est la mitochondrie. Les électrons de la chaîne respiratoire mitochondriale convertissent l'oxygène moléculaire en l'anion superoxyde (O·̱2), une espèce radicalaire modérément réactive, capable de produire le peroxyde d'hydrogène (H2O2) qui peut, à son tour, produire le radical hydroxyle OH·, par interaction avec les métaux lourds et, en particulier, le fer. Des productions locales et régulées de formes réactives de l'oxygène sont aussi assurées par un certain nombre de peroxydases. Les défenses anti-oxydantes de la cellule comprennent des enzymes telles que la superoxyde dismutase (SOD), la catalase et la glutathion peroxydase (Gpx) qui convertissent les formes réactives de l'oxygène en espèces moléculaires moins réactives. Une autre stratégie anti-oxydante mise en oeuvre par la cellule est le contrôle de la disponibilité du fer libre intracellulaire. L'existence même de fer libre dans le cytoplasme a fait l'objet de nombreuses controverses, au point que ce pool a été comparé au monstre du Loch Ness. Cependant, un faisceau d'arguments permet de proposer qu'il existe dans le cytosol des cellules un pool de fer redox-actif, faiblement lié à des composés de bas poids moléculaire, facilement « chélatable », représentant moins de 5 % du fer cellulaire total, présent à des concentrations de l'ordre de 100 nM à 1 µM suivant les cellules [1] et jouant un rôle dans la régulation post-transcriptionnelle d'un certain nombre de gènes codant pour des protéines de transport et de stockage du fer [2]. Ce fer libre (LIP, labile iron pool) réagit avec H2O2 suivant la réaction de Fenton pour former le radical hydroxyle OH· :
Fe2+ + H2O2 → Fe3+ + OH· + OH–.
Le radical OH·, nécessaire mais dangereux
Ce radical OH· est l'espèce radicalaire la plus toxique dans les cellules, capable de diffuser à travers les membranes cellulaires et d'engendrer des réactions de peroxydation lipidique, et d'oxydation des protéines, des hydrates de carbone et de l'ADN. Ces réactions radicalaires sont à la fois un danger pour la cellule, mais aussi une nécessité car elles participent à la régulation des voies de transduction et au contrôle de l'équilibre entre la vie et la mort de la cellule.
Ainsi, l'activation de l'apoptose induite par le tumor necrosis factor α (TNFα) dépend de la production des formes réactives de l'oxygène [3] qui viennent activer la voie des c-Jun N-terminal kinases (JNK), des médiateurs de la mort cellulaire programmée. Mais le TNFα est aussi un inducteur de NFκB (nuclear factor κB), qui va intervenir pour inhiber l'apoptose en bloquant l'activation des JNK. Cette activité anti-apoptotique des facteurs de transcription de la famille NFκB/Rel est cruciale pour l'immunité, la réponse inflammatoire et l'oncogenèse. L'activation de NFκB permet d'inhiber l'apoptose induite par le TNFα, principalement en réprimant la cascade des JNK. Plusieurs gènes cibles de NFκB contribuent à la répression de la cascade des JNK, mais une étude publiée dans Cell démontre que l'activation de la sous-unité H de la ferritine (HFt) permet le contrôle de la production des formes réactives de l'oxygène et l'inhibition de l'apoptose induite par le TNFα [4]. La notion d'un rôle protecteur de HFt vis-à-vis du stress oxydatif n'est pas nouvelle, et plusieurs travaux ont montré que la surexpression de la HFt dans différents modèles cellulaires protège contre le stress induit par H2O2 [5-8]. Dans l'article publié dans Cell, les auteurs vont plus loin en démontrant que la HFt est le médiateur essentiel des activités anti-apoptotiques de NFκB.
Le rôle chélateur de la ferritine
La ferritine est le chélateur naturel du fer dans les cellules, permettant de séquestrer rapidement le fer sous une forme facilement disponible, non réactive, et de constituer des réserves à long terme. Cette protéine est très conservée dans le monde du vivant, puisque des formes analogues de ferritine existent chez les bactéries, les champignons, les plantes, les vertébrés et les invertébrés. Seule la levure semble pouvoir se passer de ferritine en stockant le fer dans la vacuole. La ferritine est une protéine hétérogène, constituée d'une coquille protéique creuse de diamètre extérieur 12-13 nm et d'un noyau ferrique pouvant contenir jusqu'à 4 000 atomes de fer au sein de la cavité centrale [9]. La coquille protéique est un hétéropolymère de 24 sous-unités, réalisée par l'assemblage en proportions variables de deux sous-unités différentes appelées H et L. Ces deux sous-unités, codées par des gènes distincts, ne présentent que 50 % d'identité de leur séquence en acides aminés, mais leur structure tridimensionnelle très conservée leur permet de se co-assembler dans un même polymère de ferritine. La sous-unité H présente une activité catalytique ferroxydase qui oxyde le Fe (II) en Fe (III)[1] et qui est nécessaire à la capture du fer par la molécule de ferritine. La létalité embryonnaire précoce des souris déficientes en HFt conforte l'importance de cette sous-unité pour la survie cellulaire [10]. La sous-unité L catalyse la formation du noyau ferrique au sein de la coquille protéique, expliquant que cette sous-unité prédomine dans les tissus impliqués dans la constitution des réserves en fer (foie, rate). La transcription du gène HFt est régulée par de multiples signaux, dont le TNFα. Chez la souris, la région 5' flanquante du gène contient deux sites de fixation de NFκB [11] responsables de l'activation de la HFt par le TNFα.
La sous-unité H de la ferritine, un agent protecteur essentiel
La stratégie de clonage fonctionnel mise en oeuvre par C.G. Pham et al. [4] a permis de montrer que la HFt est capable de bloquer l'apoptose induite par le TNFα dans des cellules relA-/-, déficientes en NFκB. La transduction de ces cellules par un vecteur rétroviral exprimant la HFt protège de la mort induite par le TNFα, au même titre que la transduction de la protéine relA. Ainsi, l'activation des caspases et la dépolarisation de la membrane mitochondriale induites par le TNFα sont inhibées lorsque les cellules relA‑/‑ sont transduites avec le vecteur exprimant la HFt. Un traitement des cellules relA‑/‑ par la N-acétyl cystéine, un puissant agent anti-oxydant, ou par un chélateur du fer, aboutit à la même protection. En revanche, la transduction d'une molécule HFt mutée au site ferroxydase, rendant la protéine incompétente vis-à-vis de la capture du fer, n'offre aucune protection contre l'apoptose induite par le TNFα. Ces résultats démontrent que la HFt peut bloquer l'apoptose induite par le TNFα et qu'elle représente un médiateur important de la fonction protectrice de NFκB. Cette inhibition implique la répression de la production des formes réactives de l'oxygène et empêche l'activation soutenue de la voie JNK. Cet effet dépend de la capacité de la HFt à chélater le fer. L'augmentation du fer libre intracellulaire ou l'inhibition de la chélation du fer par HFt, en interférant avec la voie des JNK, pourrait représenter un moyen de dissocier l'effet anti-apoptotique de l'effet anti-inflammatoire de NFκB.
Figure 1
Rôle anti-apoptotique de la sous-unité H de la ferritine.
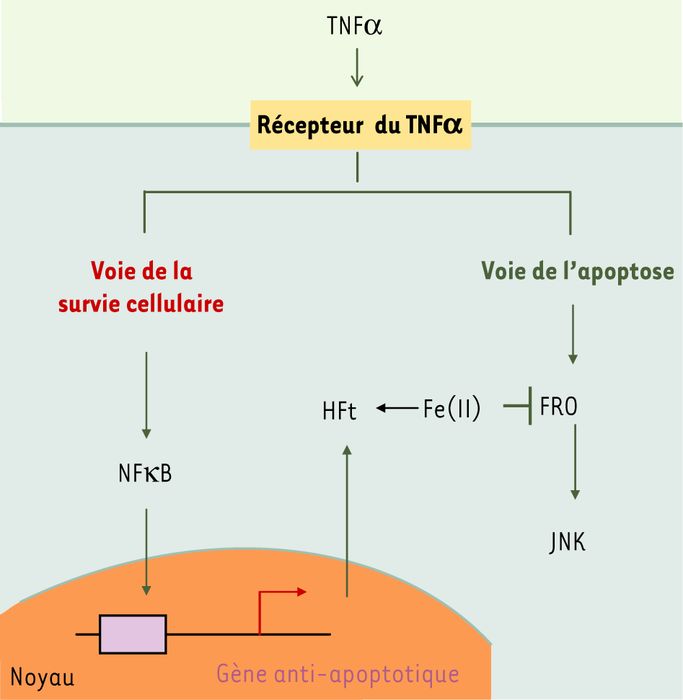
La fixation du tumor necrosis factor α (TNFα) sur son récepteur entraîne l'activation de l'apoptose, mais active aussi une voie de signalisation anti-apoptotique favorisant la survie cellulaire. Cette voie implique les facteurs de transcription de la famille NFκB qui stimulent la transcription de gènes cibles ayant un rôle protecteur, dont celui codant pour la sous-unité H de la ferritine (HFt). Celle-ci, de par sa capacité à chélater le fer libre intracellulaire, bloque la formation des formes réactives de l'oxygène (FRO) nécessaires à l'activation des c-Jun N-terminal kinases (JNK) et à l'exécution du programme de mort cellulaire.
Appendices
Note
-
[1]
Il n'est pas équivalent d'écrire Fe(II) et Fe2+. La première écriture évoque uniquement la valence du fer, indépendamment de la présence éventuelle de ligand, alors que la deuxième (Fe2+) suppose que le fer est à l'état libre ionisé.
Références
- 1. Kakhlon O, Cabantchik ZI. The labile iron pool : characterization, measurement, and participation in cellular processes. Free Radic Biol Med 2002 ; 33 : 1037-46.
- 2. Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts : molecular control of mammalian iron metabolism. Cell 2004 ; 117 : 285-97.
- 3. Varfolomeev EE, Ashkenazi A. Tumor necrosis factor : an apoptosis JuNKie? Cell 2004 ; 116 : 491-7.
- 4. Pham CG, Bubici C, Zazzeroni F, et al. Ferritin heavy chain upregulation by NF-κB inhibits TNFα-induced apoptosis by suppressing reactive oxygen species. Cell 2004 ; 119 : 529-42.
- 5. Kakhlon O, Gruenbaum Y, Cabantchik ZI. Repression of ferritin expression increases the labile iron pool, oxidative stress, and short-term growth of human erythroleukemia cells. Blood 2001 ; 97 : 2863-71.
- 6. Epsztejn S, Glickstein H, Picard V, et al. H-ferritin subunit overexpression in erythroid cells reduces the oxidative stress response and induces multidrug resistance properties. Blood 1999 ; 94 : 3593-603.
- 7. Lipinski P, Drapier JC, Oliveira L, et al. Intracellular iron status as a hallmark of mammalian cell susceptibility to oxidative stress : a study of L5178Y mouse lymphoma cell lines differentially sensitive to H2O2. Blood 2000 ; 95 : 2960-6.
- 8. Levi S, Corsi B, et al. Role of iron and ferritin in TNFα-induced apoptosis in HeLa cells. FEBS Lett 2003 ; 537 : 187-92.
- 9. Arosio P, Levi S. Ferritin, iron homeostasis, and oxidative damage. Free Radic Biol Med 2002 ; 33 : 457-63.
- 10. Ferreira C, Bucchini D, Martin ME, et al. Early embryonic lethality of H ferritin gene deletion in mice. J Biol Chem 2000 ; 275 : 3021-4.
- 11. Kwak EL, Larochelle DA, Beaumont C, et al. Role for NF-κB in the regulation of ferritin H by tumor necrosis factor-α. J Biol Chem 1995 ; 270 : 15285-93.
List of figures
Figure 1
Rôle anti-apoptotique de la sous-unité H de la ferritine.
La fixation du tumor necrosis factor α (TNFα) sur son récepteur entraîne l'activation de l'apoptose, mais active aussi une voie de signalisation anti-apoptotique favorisant la survie cellulaire. Cette voie implique les facteurs de transcription de la famille NFκB qui stimulent la transcription de gènes cibles ayant un rôle protecteur, dont celui codant pour la sous-unité H de la ferritine (HFt). Celle-ci, de par sa capacité à chélater le fer libre intracellulaire, bloque la formation des formes réactives de l'oxygène (FRO) nécessaires à l'activation des c-Jun N-terminal kinases (JNK) et à l'exécution du programme de mort cellulaire.