Abstracts
Résumé
Si l’étude des causes génétiques du cancer (mutations, amplification ou perte de matériel chromosomique, translocations récurrentes) a longtemps occupé le devant de la scène, l’explosion récente des connaissances sur les acteurs moléculaires et les mécanismes sous-jacents qui, en modulant la structure de la chromatine, contrôlent l’expression des gènes a révélé le rôle prépondérant joué par des modifications épigénétiques dans le déclenchement et la progression de nombreuses maladies, en particulier des cancers. De plus, contrairement aux modifications génétiques, les modifications épigénétiques sont dynamiques et réversibles. La caractérisation d’inhibiteurs spécifiques de certains effecteurs épigénétiques a ouvert une nouvelle voie thérapeutique, la thérapie épigénétique, qui semble très prometteuse, certaines molécules étant déjà en essais cliniques.
Summary
Epigenetics is defined as « the study of mitotically and/or meiotically heritable changes in gene expression that cannot be explained by changes in the DNA sequence ». Setting up the epigenetic program is crucial for correct development and its stable inheritance throughout its lifespan is essential for the maintenance of the tissue- and cell-specific functions of the organism. For many years, the genetic causes of cancer have hold centre stage. However, the recent wealth of information about the molecular mechanisms which, by modulating the chromatin structure, can regulate gene expression has high-lighted the predominant role of epigenetic modifications in the initiation and progression of numerous pathologies, including cancer. The nucleosome is the major target of these epigenetic regulation mechanisms. They include a series of tightly interconnected steps which starting with the setting (« writing ») of the epigenetic mark till its «reading» and interpretation will result in long-term gene regulation. The major epigenetic changes associated with tumorigenesis are aberrant DNA methylation of CpG islands located in the promoter region of tumor suppressor gene, global genomic hypomethylation and covalent modifications of histone N-terminal tails which are protruding out from the nucleosome core. In sharp contrast with genetic modifications, epigenetic modifications are highly dynamic and reversible. The characterization of specific inhibitors directed against some key epigenetic players has opened a new and promising therapeutic avenue, the epigenetic therapy, since some inhibitors are already used in clinical trials.
Article body
L’épigénétique est définie comme « l’étude des modifications de l’expression des gènes qui sont transmissibles lors de la mitose et/ou la méiose, mais ne découlent pas de modifications dans la séquence de l’ADN » [1]. La mise en place du programme épigénétique est cruciale au cours du développement, et sa stabilité est essentielle pour le maintien des fonctions de chaque type cellulaire au cours de la vie d’un organisme [2]. Les nucléosomes, unité de base du matériel génétique sont, au niveau de leur composition comme de leur arrangement, la cible principale de mécanismes de régulation épigénétiques. Ceux-ci mettent en oeuvre un ensemble complexe d’étapes étroitement interconnectées et dépendantes qui, depuis le dépôt (« écriture ») des marques épigénétiques jusqu’à leur « lecture » et leur interprétation, permettront de moduler l’activité transcriptionnelle des gènes [3-7]. Des changements épigénétiques majeurs sont associés à la cancérogenèse, notamment des méthylations aberrantes de l’ADN au niveau de dinucléotides CpG et des modifications post-traductionnelles des extrémités aminoterminales des histones faisant saillie hors du nucléosome.
Hyperméthylation locale et hypométhylation globale : le paradoxe de la méthylation dans les cellules cancéreuses
Une perte de méthylation au niveau de dinucléotides CpG, seule séquence pouvant être méthylée dans le génome des mammifères, et une diminution globale du taux de méthylcytosine furent les premières modifications épigénétiques identifiées dans les cellules cancéreuses au début des années 1980 [8]. Quelques années plus tard, une hyperméthylation spécifique du gène de la calcitonine, un marqueur du cancer du poumon à petites cellules, fut mise en évidence et corrélée à son extinction transcriptionnelle. Cette observation fut ensuite étendue au gène Rb, puis à de nombreux autres gènes suppresseurs de tumeur [5, 6].
Méthylation des motifs CpG
Les dinucléotides CpG sont répartis de façon non uniforme dans le génome, avec une sous-représentation générale, mais un enrichissement dans de courtes régions, appelées îlots CpG, positionnées au niveau du promoteur et/ou du premier exon de plus de 60 % des gènes humains [9].
Les profils de méthylation sont mis en place très tôt au cours du développement, par l’action concertée d’au moins trois enzymes, les méthyltransférases de l’ADN (DNMT1, ‑3a et ‑3b) [10, 11]. L’inactivation des gènes de ces enzymes chez la souris a montré qu’ils jouaient un rôle essentiel pour le développement normal. Un modèle a été proposé, dans lequel DNMT3a et -3b établissent les profils de méthylation au cours du développement (« méthylases de novo »), tandis que DNMT1 est considérée comme une « méthylase de maintenance », responsable de la transmission du profil de méthylation de la cellule mère aux cellules filles au cours de la réplication de l’ADN. Cependant, le rôle de DNMT1 comme seule enzyme de maintenance a récemment été remis en cause par des expériences d’invalidation génique dans des lignées de cellules colorectales humaines [12] : en effet, l’hyperméthylation de certains gènes, comme le gène suppresseur de tumeur p16INK4a, est maintenue dans des cellules Dnmt1-/-, et seule la double inactivation de Dnmt1 et de Dnmt3b permet sa réactivation [13].
Une faible augmentation de l’activité ADN-méthyltransférase est observée dans les cellules transformées, mais elle ne semble pouvoir expliquer, à elle seule, ni les méthylations aberrantes, ni surtout le ciblage de certains promoteurs. Cette spécificité pourrait être le fait de facteurs de trancription, qui recruteraient les DNMT au niveau du promoteur de leurs gènes cibles, comme dans le cas de la protéine de fusion PML-RARα (promyelocytic leukemia-récepteur α de l’acide rétinoïque) impliquée dans des leucémies promyélocytaires aiguës [14].
Les CpG méthylés sont reconnus par un motif protéique conservé, d’environ 70 acides aminés, appelé MBD (methyl-CpG binding domain). Ce motif est porté par une famille de protéines dont les principaux membres, MeCP2, MBD1, MBD2, MBD3 et MBD4, ne se lient cependant pas tous aux CpG méthylés [15] ; par ailleurs, des auteurs ont également décrit une activité d’ADN-déméthylase pour MBD2, même si cette observation fait l’objet d’une controverse. Enfin, Kaiso, un facteur de transcription associant un domaine BTB/POZ (broad complex, tramtrack and bric a brac/poxvirus and zinc finger) à des doigts de zinc de type C2H2, mais ne possédant pas de domaine MBD, s’est récemment révélé capable de fixer des CpG méthylés.
Une caractéristique commune des protéines à domaine MBD, comme de Kaiso, est de pouvoir recruter, au niveau des CpG méthylés, des complexes ayant des activités de modifications post-traductionnelles des histones (désacétylation et méthylation) entraînant une extinction transcriptionnelle (Figure 1). Dans les cellules cancéreuses (où, rappelons-le, l’hyperméthylation de certains gènes s’inscrit de façon paradoxale dans un contexte d’hypométhylation globale du génome de ces cellules), l’hyperméthylation des CpG entraîne donc une extinction transcriptionnelle, par modification épigénétique, de gènes suppresseurs de tumeur impliqués dans certaines fonctions essentielles pour la cellule, comme le contrôle du cycle cellulaire, la réparation des dommages de l’ADN et l’apoptose, à l’exception notable de la méthylation de l’ADN (Tableau I). Concernant cette dernière, si aucun lien causal direct n’a pu être établi entre l’induction de cancers et la « machinerie de méthylation », des modifications de celle-ci ont néanmoins été impliquées dans certaines maladies : ainsi, le syndrome ICF (immunodeficiency, chromosomal instability and facial anomalies), caractérisé par une perte de méthylation des centromères et une grande instabilité chromosomique, mais au cours duquel on n’observe pas de prédisposition au cancer, est lié à des mutations inactivantes de DNMT3b [16]. De même, des mutations de MeCP2 sont responsables d’un désordre neurologique, le syndrome de Rett, essentiellement observé chez les filles [17].
Figure 1
Aspects de la chromatine au niveau de gènes suppresseurs de tumeur, dans les situations normale et tumorale.

La plupart des gènes présentent, au niveau de leur promoteur et/ou de leur premier exon, un nombre élevé de dinucléotides CpG regroupés dans des zones nommées îlots CpG. Dans une situation normale, la plupart des CpG en dehors des îlots sont méthylés (cercles noirs), tandis que les CpG des îlots ne sont pas méthylés (cercles blancs). Dans cette région promotrice, l’acétylation des histones permet de maintenir la chromatine dans un état « relâché », accessible aux complexes de transcription constitués notamment de facteur de transcription (FT), de protéines acétylant les histones (HAT) et de co-activateurs transcriptionnels (CA). Dans une situation tumorale, les profils de méthylation sont inversés, avec une hypométhylation des CpG répartis le long du génome et dans les régions codantes, et une hyper-méthylation des îlots CpG au niveau des promoteurs. Ces CpG méthylés sont reconnus par des protéines à domaine MBD (methyl-CpG binding domain), responsables du recrutement, notamment, des enzymes de modifications post-traductionelles des histones (désacétylation [HDAC] et méthylation), des protéines responsables de la méthylation de l’ADN (DNMT) et des corépresseurs transcriptionnels (CR). Cette zone devient alors inaccessible aux complexes de transcription, empêchant ainsi l’expression du gène.
Tableau I
Gènes suppresseurs de tumeurs communément inactivés lors de la tumorigenèse.
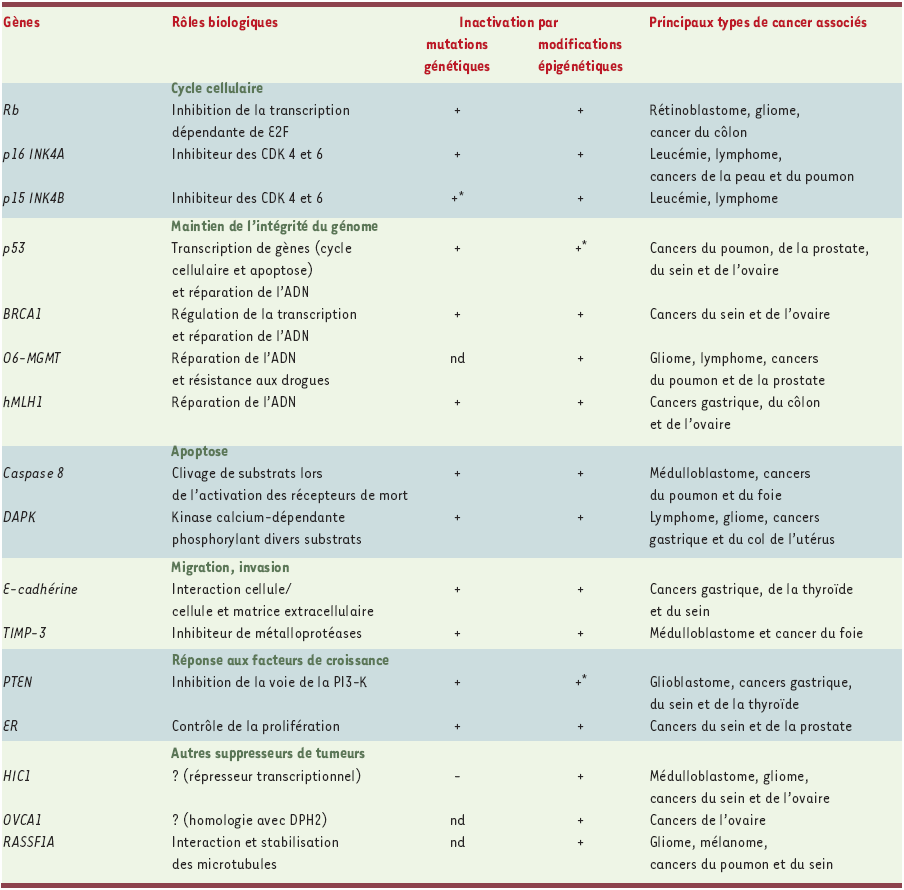
Les gènes suppresseurs de tumeurs les plus souvent inactivés par des modifications épigénétiques sont représentés dans ce tableau, ainsi que les principaux types de cancers associés à ces inactivations. Lors de la cancérisation, la perte d’expression d’un gène peut être due soit à des pertes d’hétérozygotie, soit à des mutations. Cependant, les travaux effectués ces dernières années permettent de démontrer que les modifications épigénétiques telles que l’hypermétylation des CpG au niveau du promoteur sont également responsables de nombreuses extinctions de l’expression. Pour certains gènes tels que p53 et PTEN, des cas d’inactivation due à une hyperméthylation n’ont été décrits que très récemment (+*) ; pour d’autres tels que 06-MGMT, OVCA1 et RASSF1, la perte d’expression résulte de modifications épigénétiques et, à ce jour, aucune mutation de ces gènes n’a été décrite (nd). Enfin, dans le cas de HIC1, l’extinction génique semble exclusivement due à l’hyperméthylation, puisque des travaux tentant de découvrir des mutations de ce gène se sont révélés négatifs (-). Rb : retinoblastoma ; CDK : cylin dependent kinase ; BRCA1 : breast cancer 1 ; O6-MGMT : 06-methyl guanine DNA methyl transferase ; HMLH1 : human mutant L homologue ; DAPK : death associated protein kinase ; TIMP-3 : tissue inhibitor of metalloproteinase 3 ; PTEN : phosphatase and tensin homologue ; PI3-K : phosphatidylinositol 3 kinase ; ER : estrogen receptor ; HIC1 : hypermethylated in cancer 1 ; OVCA1 : ovarian cancer 1 ; RASSF1A : ras effector homologue ; DPH2 : diphtamide biosynthesis protein 2.
Gènes cibles de l’hyperméthylation
S’il semble établi que l’hyperméthylation de certains gènes est une cause plutôt qu’une conséquence de la tumorigenèse, l’observation d’une telle modification épigénétique implique-t-elle forcément que le gène affecté soit un gène suppresseur de tumeur ? L’utilisation de modèles animaux peut permettre de répondre à cette question.
Le gène HIC1 (hypermethylated in cancer 1), codant un répresseur transcriptionnel, a été isolé dès 1995 au voisinage d’un marqueur microsatellitaire, situé en 17p13.3, fréquemment hyperméthylé dans de nombreux cancers humains. Cependant, sa fonction de gène suppresseur de tumeur n’a été établie que très récemment, grâce à l’obtention de souris hétérozygotes qui, ayant une copie du gène invalidée par recombinaison homologue (Hic1+/-), développent tardivement dans leur vie des tumeurs spontanées [18].
Les gènes soumis à l’empreinte parentale ((→) m/s 2005, n° 4, p. 390) constituent un autre groupe de gènes porteurs de méthylations aberrantes au cours de la tumorigenèse. Une région située sur le chromosome 11p15, et contenant une dizaine de gènes soumis à empreinte, est impliquée dans le syndrome de Beckwith-Wiedemann, une maladie qui se traduit par des anomalies de formation d’organes et une prédisposition à certains cancers chez l’enfant, notamment à la tumeur de Wilms.
Bien que certains îlots CpG soient hyperméthylés, le génome des cellules cancéreuses se caractérise par une hypométhylation génomique globale qui peut avoir plusieurs implications fonctionnelles. Tout d’abord, en parfaite opposition avec l’inactivation de gènes suppresseurs par hyperméthylation, elle peut toucher des îlots CpG méthylés dans les tissus normaux, et donc aboutir à la réactivation de certains gènes. Mais, l’hypométhylation est surtout particulièrement importante au niveau de séquences satellites péricentromériques, ce qui pourrait prédisposer l’ADN à des cassures ou des remaniements chromosomiques. De tels événements sont fréquemment observés au cours de plusieurs cancers, notamment dans le cas des tumeurs de Wilms, qui présentent des translocations chromosomiques non équilibrées au niveau de l’ADN péricentromérique des chromosomes 1 et 16. Par ailleurs, on peut noter que l’hypométhylation génomique globale augmente avec l’âge, ce qui pourrait en partie expliquer l’incidence accrue des cancers chez les personnes âgées.
Tous ces résultats montrent bien l’importance de cette modification épigénétique par méthylation de l’ADN dans l’apparition des cancers ; ils indiquent également qu’un équilibre subtil entre hyperméthylation et hypométhylation doit être maintenu à l’intérieur de la cellule.
Un code épigénétique complexe : les modifications covalentes des histones
Les extrémités aminoterminales des histones, qui font saillie à l’extérieur du nucléosome, sont la cible privilégiée de modifications épigénétiques responsables de l’état transcriptionnel de la chromatine selon le « code histone » [19] ((→) m/s 2005, n° 4, p. 384). Si de nombreuses modifications covalentes ont été mises en évidence, les plus étudiées restent aujourd’hui l’acétylation et la méthylation des résidus lysine.
Le niveau d’acétylation est la résultante de l’activité de deux familles d’enzymes antagonistes : les histone-acétyltransférases (HAT) et les histone-désacétylases (HDAC). En désacétylant les résidus lysine, les HDAC permettent de passer d’une chromatine « permissive » pour la transcription à une chromatine « réprimée », cet effet pouvant être renforcé par l’intervention ultérieure d’histone-méthyltransférases (HMT) ciblant les mêmes résidus (Figure 2).
Figure 2
Modifications épigénétiques et réactivation pharmacologique.
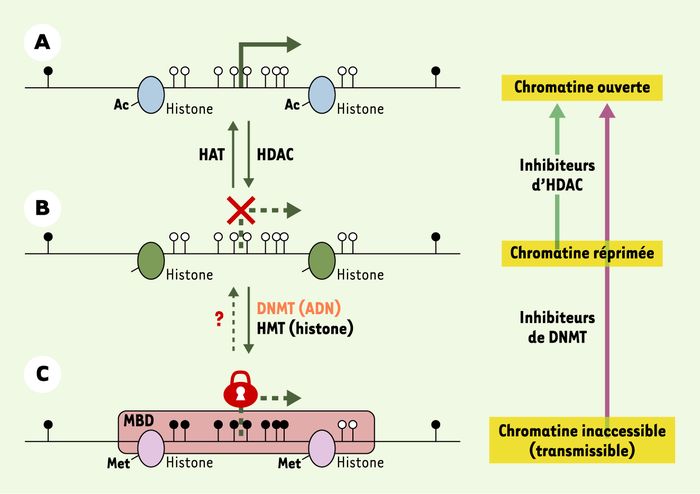
La désacétylation des résidus lysine des histones par les HDAC (histone-désacétylases) permet de passer d’une chromatine « ouverte » (A), accessible pour la transcription, à une chromatine « réprimée » (B), dans laquelle la transcription est inhibée. Néanmoins, une extinction génique transmissible lors de la mitose et/ou de la méiose est le résultat d’un « verrouillage » (C) encore plus important de la chromatine, caractérisé par : (1) la présence de modifications des histones, avec une désacétylation (par des HADC) accompagnée d’une hyperméthylation par des HMT (histone-méthyltransférases) des résidus lysines ; (2) une méthylation des îlots CpG par des DNMT (méthyltransférases de l’ADN) ; et (3) la fixation de protéines à MBD (methyl CpG binding domain) sur les CpG méthylés. Alors que plusieurs histone-acétyltransférases (HAT), telles que CBP (creb binding protein) et P300, ont été décrites, la première histone déméthylase vient juste d’être découverte et l’existence d’une déméthylase de l’ADN reste un sujet de controverse. Dans le cadre d’une thérapie épigénétique antitumorale, la réactivation de la transcription des gènes inactivés est possible par l’utilisation combinée d’agents modulant l’acétylation des histones et la méthylation de l’ADN. Ainsi, les inhibiteurs d’HDAC réactivent la transcription de gènes dont les promoteurs sont déméthylés, mais pas celle des gènes dont les promoteurs sont hyperméthylés. En revanche, les inhibiteurs de DNMT permettent de déverrouiller la transcription en déméthylant l’ADN au niveau des îlots CpG de la région promotrice, et de modifier la structure de la chromatine, d’où une réexpression des gènes.
Si l’acétylation est clairement associée à l’activation transcriptionnelle et la désacétylation à la répression, le rôle de la méthylation semble en revanche plus complexe. En effet, la conséquence fonctionnelle de cette modification épigénétique est variable : ainsi, la méthylation de la lysine 4 de l’histone H3 est corrélée à une activation transcriptionnelle, tandis que celle de la lysine 9 est corrélée à une répression. De plus, le « code histone » se révèle beaucoup plus complexe en ce qui concerne la méthylation, les lysines pouvant être mono-, di- ou triméthylées [20]. Enfin, on peut noter que la première histone déméthylase, spécifique de la lysine 4 de l’histone H3, n’a été caractérisée que très récemment ((→) m/s 2003, n° 4, p. 384).
Les histone-acétyltransférases (HAT) comme CBP (creb binding protein) et P300, co-activateurs trancriptionnels de diverses protéines, sont la cible de nombreuses translocations chromosomiques associées à des leucémies. De plus, des mutations ponctuelles du gène p300, souvent accompagnées de l’inactivation du second allèle, sont retrouvées dans des tumeurs solides. Enfin, des mutations germinales de CBP sont associées au syndrome de Rubinstein-Taybi, caractérisé par un retard mental, des anomalies faciales et une prédisposition au cancer chez l’enfant [21]. De façon inattendue, aucune corrélation aussi nette avec des maladies n’a pu être établie dans le cas des histone-désacétylases (HDAC).
Une douzaine d’histone-méthyltransférases (HMT) spécifiques des lysines, qu’elles soient impliquées dans l’activation ou la répression transcriptionnelle, ont été liées plus au moins directement au cancer [22]. Le premier exemple décrit fut celui du locus ALL1/MLL1 (acute lymphoid leukemia/myeloid/lymphoid or mixed-lineage leukemia) localisé en 11q23, une région impliquée dans une quarantaine de translocations chromosomiques associées à environ 80 % des leucémies aiguës chez l’enfant. Une caractéristique commune des protéines de fusion obtenues est l’élimination de l’extrémité carboxyterminale de MLL1, contenant le domaine SET (su[var]3-9, enhancer-of-zeste, trithorax), capable de méthyler la lysine 4 de l’histone H3. MLL1 est l’orthologue humain du gène trithorax de drosophile, prototype du groupe Trithorax (Trx-G) dont la fonction, antagoniste de celle des membres du groupe Polycomb (Pc-G), est la régulation positive de l’expression des gènes au cours du développement. Des protéines Polycomb ont également été caractérisées chez les mammifères, et plusieurs d’entre elles impliquées dans la tumorigenèse : c’est notamment le cas de l’histone-méthyltransférase EZH2, ciblant principalement la lysine 27 de l’histone H3. EZH2 est surexprimée dans de nombreux cancers, en raison d’une perte de son contrôle par la voie Rb-E2F ; cette surexpression est un marqueur de mauvais pronostic pour le cancer de la prostate et du sein [23].
Histone-acétyltransférases et histone-désacétylases coopèrent avec des complexes de remodelage de la chromatine utilisant l’énergie issue de l’hydrolyse de l’ATP pour modifier l’accessibilité de l’ADN nucléosomique. Selon la sous-unité ATPasique impliquée, ces complexes ont été regroupés en trois sous-familles dont deux, SWI/SNF (faulty mating-type switching/sucrose non fermenting) et Mi-2/NuRD (mi-2/nucleosome remodelling and deacetylation), jouent un rôle important dans la tumorigenèse [24]. Ainsi, BRG1 (brahma/swi2-related gene 1), l’ATPase de l’un des complexes SWI/SNF humains, est mutée dans de nombreuses tumeurs ; de plus, son invalidation chez la souris a montré que BRG1 est un gène suppresseur de tumeur haplo-insuffisant, puisque les souris hétérozygotes brg1+/-, si elles sont viables et fertiles, développent des tumeurs vers l’âge de 16 mois. SNF5, l’un des constituants des complexes SWI/SNF, est inactivé dans de nombreux types de tumeurs, en particulier les tumeurs malignes rhabdoïdes [24]. Enfin, la perte d’expression de MTA3 (metastasis associated gene 3), un constituant spécifique de tissu du complexe Mi-2/NURD, a récemment été impliquée dans l’agressivité et le pouvoir métastatique des cancers du sein [25].
Les modifications épigénétiques des histones, grâce à une série d’étapes étroitement dépendantes, permettent de passer d’une chromatine transcrip- tionellement active à une chromatine réprimée, d’abord de façon réversible (acétylation/désacétylation), puis d’une façon plus stable (méthylation). Récemment, des études réalisées chez les plantes et les champignons ont clairement établi une dépendance étroite et réciproque entre la méthylation des histones et celle de l’ADN : cette boucle de régulation permet de propager et de maintenir, de façon plus ou moins stable et localisée, une structure chromatinienne réprimée de type hétérochromatine au niveau de gènes de l’euchromatine, y compris des gènes suppresseurs de tumeur [26].
Une nouvelle approche thérapeutique : la thérapie épigénétique
Contrairement aux causes génétiques du cancer qui affectent la séquence de l’ADN, les modifications épigénétiques sont réversibles, ce qui ouvre des perspectives prometteuses en thérapie. Deux grandes familles d’inhibiteurs sont disponibles, spécifiques des méthyltransférases de l’ADN (DNMT) ou des histone-désacétylases (HDAC) (Figure 2) [3].
La 5-azacytidine, la 5-aza-2’-désoxycytidine et leurs analogues sont des inhibiteurs des DNMT, qu’elles piègent en s’intégrant dans l’ADN à la place de résidus cytosine, permettant ainsi la réexpression de gènes suppresseurs de tumeur éteints par l’hyperméthylation. Plusieurs dérivés modifiés pour diminuer la cytotoxicité de ces composés et augmenter leur stabilité en solution aqueuse ont été utilisés en tests cliniques, et ont donné des résultats significatifs dans le traitement de différents types de leucémie [27].
Les inhibiteurs d’HDAC sont quant à eux regroupés en quatre groupes selon leur classe structurale. Parmi les plus connus, on peut citer des acides gras à courte chaîne, comme le butyrate, des acides hydroxamiques, comme la trichostatine A (TSA), et des tétrapeptides cycliques, comme la trapoxine. Dans le cas de leucémies promyélocytaires aiguës résistantes au traitement par l’acide rétinoïque (ATRA), l’utilisation conjointe d’inhibiteurs d’HDAC et d’ATRA induit des rémissions [14].
La démonstration du dialogue entre la méthylation des histones et celle de l’ADN [4, 26], ainsi que les nombreux résultats expérimentaux obtenus sur diverses lignées cellulaires [28] orientent les protocoles thérapeutiques vers une utilisation conjointe de ces deux types d’inhibiteurs, ou en complément de thérapies classiques. En dépit de nombreux problèmes encore à résoudre, notamment de toxicité et de spécificité, et même si tous les mécanismes moléculaires ne sont pas encore totalement élucidés [29], cette nouvelle approche thérapeutique est sans aucun doute promise à un bel avenir.
Conclusions
L’importance cruciale des modifications épigénétiques au cours du développement normal et leur implication dans de nombreuses maladies ne font plus aucun doute. Un champ fantastique de recherche a ainsi été ouvert, même si de nombreux mécanismes et acteurs moléculaires doivent encore être découverts. Outre son intérêt théorique dans le décryptage des mécanismes fondamentaux de la régulation de l’expression des gènes, l’épigénétique ouvre également des perspectives fascinantes pour l’utilisation de nouvelles approches thérapeutiques. Ainsi, ces dernières permettraient de restaurer l’expression des multiples gènes hyperméthylés au sein d’un même type de cancer. Par ailleurs, il a été démontré que le nombre de CpG méthylés augmentait avec l’âge, contribuant en partie au développement de maladies chroniques : dans ce cadre, l’utilisation des thérapies épigénétiques pourrait être également envisagée. Toutefois, bien que plusieurs agents inhibant la méthylation aient été découverts à ce jour, la plupart ne sont pas spécifiques des méthyltransférases de l’ADN. Les récents progrès concernant la compréhension des phénomènes épigénétiques permettent d’envisager dès à présent des approches plus rationnelles, notamment au niveau de la modélisation moléculaire d’agents ciblant spécifiquement ces modifications épigénétiques.
Appendices
Remerciements
Sophie Deltour et Valérie Chopin remercient respectivement l’EMBO et la Ligue Nationale contre le Cancer pour leur soutien financier.
Références
- 1. Wollfe AP, Matzke MA. Epigenetics : regulation through repression. Science 1999 ; 286 : 481-6.
- 2. Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Genet 2002 ; 3 : 662-73.
- 3. Egger G, Liang G, Aparicio A, et al. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004 ; 429 : 457-63.
- 4. Lund AH, van Lohuizen M. Epigenetics and cancer. Genes Dev 2004 ; 18 : 2315-35.
- 5. Jones PA, Baylin SB. The fundamental roles of epigenetic events in cancer. Nat Rev Genet 2002 ; 3 : 415-28.
- 6. Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 2003 ; 349 : 2042-54.
- 7. Hake SB, Xiao A, Allis CD. Linking the epigenetic language of covalent histone modifications to cancer. Br J Cancer 2004 ; 90 : 761-9.
- 8. Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer 2004 ; 4 : 143-53.
- 9. Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002 ; 16 : 6-21.
- 10. Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet 2000 ; 9 : 2395-402.
- 11. Fuks F. Les méthyltransférases de l’ADN : du remodelage de la chromatine au cancer. Med Sci (Paris) 2003 ; 19 : 477-80.
- 12. Ting AH, Jair KW, Suzuki H, et al. CpG island hypermethylation is maintained in human colorectal cancer cells after RNAi-mediated depletion of DNMT1. Nat Genet 2004 ; 36 : 582-4.
- 13. Rhee I, Bachman KE, Park BH, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 2002 ; 416 : 552-6.
- 14. Claus R, Lübbert M. Epigenetic target in hematopietic malignancies. Oncogene 2003 ; 22 : 6489-96.
- 15. Filion G, Defossez PA. Les protéines se liant à l’ADN méthylé : interprètes du code épigénétique. Med Sci (Paris) 2004 ; 20 : 7-8.
- 16. Xu GL, Bestor TH, Bourc’his D, et al. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999 ; 402 : 187-91.
- 17. Kriaucionis S, Bird A. DNA methylation and Rett syndrome. Hum Mol Genet 2003 ; 12 : 221-7.
- 18. Chen WY, Zeng X, Carter MG, et al. Heterozygous disruption of Hic1 predisposes mice to a gender-dependent spectrum of malignant tumors. Nat Genet 2003 ; 33 : 197-202.
- 19. Jenuwein T, Allis CD. Translating the histone code. Science 2001 ; 293 : 1074-80.
- 20. Kouzarides T. Histone methylation in transcriptional control. Curr Opin Genet Dev 2002 ; 12 : 198-209.
- 21. Iyer NG, Ozdag H, Caldas C. p300/CBP and cancer. Oncogene 2004 ; 23 : 4225-31.
- 22. Schneider R, Bannister A, Kouzarides T. Unsafe SETs : histone lysine methyltransferases and cancer. Trends Biochem Sci 2002 ; 27 : 396-402.
- 23. Cao R, Zhang Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr Opin Genet Dev 2004 ; 14 : 155-64.
- 24. Roberts CWM, Orkin SH. The SWI/SNF complex-chromatin and cancer. Nat Rev Cancer 2004 ; 4 : 133-42.
- 25. Fujita N, Jaye DL, Kajita M, et al. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell 2003 ; 113 : 207-19.
- 26. Fahrner JA, Baylin, SB. Heterochromatin : stable and unstable invasions at home and abroad. Genes Dev 2003 ; 17 : 1805-12.
- 27. Villar-Garea A, Esteller M. DNA demethylating agents and chromatin-remodelling drugs : which, how and why. Curr Drug Metab 2003 ; 4 : 11-31.
- 28. Bachman KE, Park BH, Rhee I, et al. Histone modifications and silencing prior to DNA methylation of a tumor suppressor gene. Cancer Cell 2003 ; 3 : 89-95.
- 29. Gius D, Cui H, Bradbury CM, et al. Distinct effects on gene expression of chemical and genetic manipulation of the cancer epigenome revealed by a multimodality approach. Cancer Cell 2004 ; 6 : 361-71.
List of figures
Figure 1
Aspects de la chromatine au niveau de gènes suppresseurs de tumeur, dans les situations normale et tumorale.
La plupart des gènes présentent, au niveau de leur promoteur et/ou de leur premier exon, un nombre élevé de dinucléotides CpG regroupés dans des zones nommées îlots CpG. Dans une situation normale, la plupart des CpG en dehors des îlots sont méthylés (cercles noirs), tandis que les CpG des îlots ne sont pas méthylés (cercles blancs). Dans cette région promotrice, l’acétylation des histones permet de maintenir la chromatine dans un état « relâché », accessible aux complexes de transcription constitués notamment de facteur de transcription (FT), de protéines acétylant les histones (HAT) et de co-activateurs transcriptionnels (CA). Dans une situation tumorale, les profils de méthylation sont inversés, avec une hypométhylation des CpG répartis le long du génome et dans les régions codantes, et une hyper-méthylation des îlots CpG au niveau des promoteurs. Ces CpG méthylés sont reconnus par des protéines à domaine MBD (methyl-CpG binding domain), responsables du recrutement, notamment, des enzymes de modifications post-traductionelles des histones (désacétylation [HDAC] et méthylation), des protéines responsables de la méthylation de l’ADN (DNMT) et des corépresseurs transcriptionnels (CR). Cette zone devient alors inaccessible aux complexes de transcription, empêchant ainsi l’expression du gène.
Figure 2
Modifications épigénétiques et réactivation pharmacologique.
La désacétylation des résidus lysine des histones par les HDAC (histone-désacétylases) permet de passer d’une chromatine « ouverte » (A), accessible pour la transcription, à une chromatine « réprimée » (B), dans laquelle la transcription est inhibée. Néanmoins, une extinction génique transmissible lors de la mitose et/ou de la méiose est le résultat d’un « verrouillage » (C) encore plus important de la chromatine, caractérisé par : (1) la présence de modifications des histones, avec une désacétylation (par des HADC) accompagnée d’une hyperméthylation par des HMT (histone-méthyltransférases) des résidus lysines ; (2) une méthylation des îlots CpG par des DNMT (méthyltransférases de l’ADN) ; et (3) la fixation de protéines à MBD (methyl CpG binding domain) sur les CpG méthylés. Alors que plusieurs histone-acétyltransférases (HAT), telles que CBP (creb binding protein) et P300, ont été décrites, la première histone déméthylase vient juste d’être découverte et l’existence d’une déméthylase de l’ADN reste un sujet de controverse. Dans le cadre d’une thérapie épigénétique antitumorale, la réactivation de la transcription des gènes inactivés est possible par l’utilisation combinée d’agents modulant l’acétylation des histones et la méthylation de l’ADN. Ainsi, les inhibiteurs d’HDAC réactivent la transcription de gènes dont les promoteurs sont déméthylés, mais pas celle des gènes dont les promoteurs sont hyperméthylés. En revanche, les inhibiteurs de DNMT permettent de déverrouiller la transcription en déméthylant l’ADN au niveau des îlots CpG de la région promotrice, et de modifier la structure de la chromatine, d’où une réexpression des gènes.
List of tables
Tableau I
Gènes suppresseurs de tumeurs communément inactivés lors de la tumorigenèse.
Les gènes suppresseurs de tumeurs les plus souvent inactivés par des modifications épigénétiques sont représentés dans ce tableau, ainsi que les principaux types de cancers associés à ces inactivations. Lors de la cancérisation, la perte d’expression d’un gène peut être due soit à des pertes d’hétérozygotie, soit à des mutations. Cependant, les travaux effectués ces dernières années permettent de démontrer que les modifications épigénétiques telles que l’hypermétylation des CpG au niveau du promoteur sont également responsables de nombreuses extinctions de l’expression. Pour certains gènes tels que p53 et PTEN, des cas d’inactivation due à une hyperméthylation n’ont été décrits que très récemment (+*) ; pour d’autres tels que 06-MGMT, OVCA1 et RASSF1, la perte d’expression résulte de modifications épigénétiques et, à ce jour, aucune mutation de ces gènes n’a été décrite (nd). Enfin, dans le cas de HIC1, l’extinction génique semble exclusivement due à l’hyperméthylation, puisque des travaux tentant de découvrir des mutations de ce gène se sont révélés négatifs (-). Rb : retinoblastoma ; CDK : cylin dependent kinase ; BRCA1 : breast cancer 1 ; O6-MGMT : 06-methyl guanine DNA methyl transferase ; HMLH1 : human mutant L homologue ; DAPK : death associated protein kinase ; TIMP-3 : tissue inhibitor of metalloproteinase 3 ; PTEN : phosphatase and tensin homologue ; PI3-K : phosphatidylinositol 3 kinase ; ER : estrogen receptor ; HIC1 : hypermethylated in cancer 1 ; OVCA1 : ovarian cancer 1 ; RASSF1A : ras effector homologue ; DPH2 : diphtamide biosynthesis protein 2.