Article body
Le fonctionnement normal d’un tissu dépend de l’apport en oxygène et en nutriments par les vaisseaux sanguins. La compréhension des mécanismes impliqués dans la formation du système vasculaire est donc devenue un objectif majeur au cours de ces dernières années. Pendant l’embryogenèse, la formation des vaisseaux se produit en deux étapes, la vasculogenèse et l’angiogenèse. La vasculogenèse correspond à l’apparition des vaisseaux primaires formant un réseau vasculaire immature. Puis ce réseau se ramifie et se stabilise lors de l’angiogenèse. À l’âge adulte, de nouveaux vaisseaux se développent également par ce processus angiogénique [1]. L’angio-_ genèse - nécessaire à la formation et au bon fonctionnement de l’organisme - peut cependant devenir délétère. Lors du développement de tumeurs solides, la néoformation de vaisseaux sanguins permet la croissance tumorale au-delà de 2 mm de diamètre. De plus, ce système vasculaire anormal favorise la dissémination des métastases dans la circulation sanguine [2]. La capacité d’une tumeur à promouvoir cette angiogenèse dépend de mutations acquises lors de la transformation des cellules.
Des formes constitutivement actives des protéines Ras ont été décrites dans un grand nombre de cancers tels que les adénocarcinomes du pancréas, du côlon ou du foie… [3]. Une fois activée, cette petite protéine G induit l’accumulation des dérivés toxiques de l’oxygène, encore appelés radicaux libres ou ROS (reactive oxygen species) et favorise la croissance tumorale [4]. Les ROS sont des facteurs importants pour le développement et l’angiogenèse des tumeurs [5]. L’activation de la protéine Ras augmente la vascularisation des tumeurs en stimulant l’expression du facteur pro-angiogénique VEGF-A (vascular endothelial growth factor-A) [6]. L’expression de VEGF-A est contrôlée par le facteur de transcription HIF-1 (hypoxia inducible factor-1). Ce facteur pallie un déficit en oxygène, appelé stress hypoxique, en activant l’expression de gènes pro-angiogéniques comme VEGF-A, érythropoïétiques tels que l’érythropoïétine, et glycolytiques comme le transporteur spécifique du glucose Glut-1 [7]. HIF-1 est un hétérodimère formé par deux sous-unités, HIF-1α et HIF-1β. Si la sous-unité β est présente constitutivement dans les cellules, la sous-unité α s’accumule dans les cellules soumises à un stress hypoxique. L’activation constitutive de Ras stabilise la sous-unité HIF-1α en conditions normoxiques. Les mécanismes moléculaires impliqués dans cette stabilisation de HIF-1α par ras n’ont été que récemment définis [8].
Notre étude décrypte, en effet, le mécanisme de stabilisation de la protéine HIF-1α par les ROS en normoxie et son impact majeur sur l’angiogenèse tumorale [8]. Il était déjà établi qu’en présence d’oxygène, HIF-1α est hydroxylée par les enzymes HIF-prolyl hydroxylases (PHD), dont l’activité dépend de l’oxygène et de deux co-facteurs, le 2-oxoglutarate et le fer ferreux (Fe2+) [9]. L’hydroxylation de HIF-1α induit son interaction avec la protéine VHL (Von Hippel-Lindau), membre d’un complexe ubiquitine-ligase de type E3 qui favorise l’ubiquitinylation et la dégradation par le protéasome de HIF-1α. Notre étude a mis en évidence que l’accumulation d’oxydants inhibe l’activité des PHD même en présence d’oxygène. Cette inhibition est due à une diminution du Fe2+ au sein du site catalytique de ces enzymes. Cette diminution résulte elle-même de l’oxydation du Fe2+ en fer ferrique (Fe3+) par le H2O2, décrite par la réaction physicochimique de Fenton (H2O2 + Fe2+ → OH. + OH- + Fe3+). Ainsi, en favorisant l’accumulation de ROS, la transformation cellulaire réduit l’activité des PHD menant à la stabilisation de HIF-1α. HIF-1 stimule alors l’expression de VEGF-A et l’angiogenèse tumorale (Figure 1). Cette étude décrit un important mécanisme de régulation des enzymes PHD contrôlant la dégradation de la protéine HIF-1α et l’angiogenèse. Ces enzymes initialement identifiées comme des « senseurs » de l’oxygène, apparaissent aussi régulées par des variations de l’équilibre redox de la cellule. En effet, l’oxydation du Fe2+ en Fe3+ en leur site catalytique inhibe l’activité des PHD. Or, de nombreuses causes physiologiques ou pathologiques, telles la transformation cellulaire ou l’infection, peuvent conduire à un déséquilibre de cet état redox et donc à un dérèglement des PHD. La découverte de ce nouveau mécanisme de régulation permettra de mieux expliquer l’accumulation de la protéine HIF-1α dans certaines maladies et de développer de nouvelles stratégies thérapeutiques.
Figure 1
Mécanisme de régulation des radicaux libres (ROS) et de l’angiogenèse par JunD.
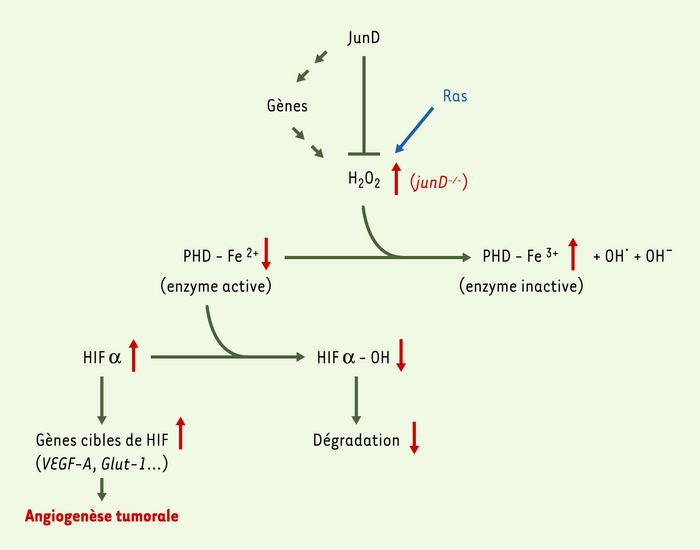
Le facteur de transcription JunD protège les cellules du stress oxydatif en réglant négativement l’expression de gènes impliqués dans la production de peroxyde d’hydrogène (H2O2) et positivement ceux impliqués dans la défense contre ce stress. La délétion du gène junD (junD-/- : flèches rouges) provoque une accumulation de H2O2. Le peroxyde d’hydrogène oxyde Fe2+ en Fe3+ au niveau du site catalytique des HIF-prolyl hydroxylases (PHD) et altère leur activité. Par conséquent, la protéine HIF-1α s’accumule et stimule la transcription du gène pro-angiogénique VEGF-A, induisant l’angiogenèse tumorale. Réciproquement, la surexpression de JunD diminue la quantité de H2O2 intracellulaire, réduisant l’effet toxique du stress oxydatif et l’angiogenèse tumorale induite par l’oncogène ras.
Notre étude a de plus révélé que JunD, membre de la famille des facteurs de transcription AP-1 (activator protein 1), réduit l’angiogenèse tumorale en limitant la production d’oxydants. La surexpression de JunD dans des fibroblastes transformés par l’oncogène ras favorise l’inversion partielle du phénotype transformé des cellules [10]. JunD possède également un effet anti-angiogénique puissant en réprimant l’expression de VEGF-A (Figure 2). Par ailleurs, l’expression de VEGF-A est stimulée dans les cellules déficientes pour le gène junD (junD-/-). Ainsi, si l’expression constitutive de JunD diminue la transcription de VEGF-A, sa délétion augmente son expression. Cet effet de balancier résulte de l’action de JunD sur les oxydants. Les cellules junD-/- accumulent des oxydants qui stabilisent HIF-1α selon le mécanisme dépendant du Fe2+ décrit plus haut ; réciproquement, la surexpression de JunD protège les cellules d’un stress oxydatif. L’effet protecteur de JunD contre le stress oxydatif revêt une importance particulière par sa capacité à réduire le développement vasculaire tumoral. Ces résultats suggèrent que JunD a un impact sur d’autres phénomènes nécessitant la formation de nouveaux vaisseaux sanguins, par exemple : ischémie, cicatrisation ou renouvellement de l’endomètre lors du cycle menstruel chez la femme. Enfin, il ouvre également de nouvelles voies d’investigation dans la compréhension des différentes maladies liées au stress oxydatif.
Figure 2
Effet anti-angiogénique de JunD.
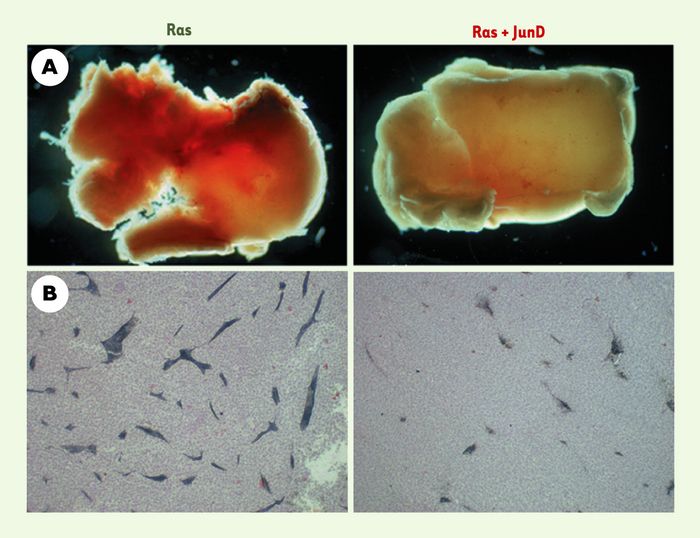
A. Tumeurs représentatives issues de l’injection de cellules cancéreuses chez des souris nude. Les tumeurs dérivées des cellules transformées par l’oncogène ras (Ras) apparaissent fortement hémorragiques et vascularisées. Au contraire, les tumeurs dérivées des cellules transformées par ras et surexprimant JunD (Ras + JunD) sont pâles et peu vascularisées. B. Analyse immunohistochimique sur coupes à l’aide d’un marqueur spécifique des cellules endothéliales (PECAM1, platelet endothelial cell adhesion molecular marker1). La surexpression de JunD diminue le nombre et la taille des vaisseaux sanguins.
Appendices
Références
- 1. Jain RK. Molecular regulation of vessel maturation.Nat Med 2003 ; 9 : 685-93.
- 2. Folkman J. What is the evidence that tumors are angiogenesis dependent ? J Natl Cancer Inst 1990 ; 82 : 4-6.
- 3. Bos JL. ras oncogenes in human cancer : a review.Cancer Res 1989 ; 49 : 4682-9.
- 4. Irani K, Xia Y, Zweier JL, et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts.Science 1997 ; 275 : 1649-52.
- 5. Brown NS,Bicknell R. Hypoxia and oxidative stress in breast cancer. Oxidative stress : its effects on the growth, metastatic potential and response to therapy of breast cancer.Breast Cancer Res 2001 ; 3 : 323-7.
- 6. Kranenburg O, Gebbink MF, Voest EE. Stimulation of angiogenesis by Ras proteins.Biochim Biophys Acta 2004 ; 1654 : 23-37.
- 7. Lee JW, Bae SH, Jeong JW, et al. Hypoxia-inducible factor (HIF-1)α : its protein stability and biological functions.Exp Mol Med 2004 ; 36 : 1-12.
- 8. Gerald D, Berra E, Frapart YM, et al. JunD reduces tumor angiogenesis by protecting cells from oxidative stress.Cell 2004 ; 118 : 781-94.
- 9. Epstein AC, Gleadle JM, McNeill LA, et al.C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation.Cell 2001 ; 107 : 43-54.
- 10. Pfarr CM, Mechta F, Spyrou G, et al. Mouse JunD negatively regulates fibroblast growth and antagonizes transformation by ras.Cell 1994 ; 76 : 747-60.
List of figures
Figure 1
Mécanisme de régulation des radicaux libres (ROS) et de l’angiogenèse par JunD.
Le facteur de transcription JunD protège les cellules du stress oxydatif en réglant négativement l’expression de gènes impliqués dans la production de peroxyde d’hydrogène (H2O2) et positivement ceux impliqués dans la défense contre ce stress. La délétion du gène junD (junD-/- : flèches rouges) provoque une accumulation de H2O2. Le peroxyde d’hydrogène oxyde Fe2+ en Fe3+ au niveau du site catalytique des HIF-prolyl hydroxylases (PHD) et altère leur activité. Par conséquent, la protéine HIF-1α s’accumule et stimule la transcription du gène pro-angiogénique VEGF-A, induisant l’angiogenèse tumorale. Réciproquement, la surexpression de JunD diminue la quantité de H2O2 intracellulaire, réduisant l’effet toxique du stress oxydatif et l’angiogenèse tumorale induite par l’oncogène ras.
Figure 2
Effet anti-angiogénique de JunD.
A. Tumeurs représentatives issues de l’injection de cellules cancéreuses chez des souris nude. Les tumeurs dérivées des cellules transformées par l’oncogène ras (Ras) apparaissent fortement hémorragiques et vascularisées. Au contraire, les tumeurs dérivées des cellules transformées par ras et surexprimant JunD (Ras + JunD) sont pâles et peu vascularisées. B. Analyse immunohistochimique sur coupes à l’aide d’un marqueur spécifique des cellules endothéliales (PECAM1, platelet endothelial cell adhesion molecular marker1). La surexpression de JunD diminue le nombre et la taille des vaisseaux sanguins.