Abstracts
Résumé
Le protéasome est la principale machinerie protéolytique de la cellule. Il est impliqué dans toutes les grandes fonctions et décisions cellulaires. On a longtemps pensé que presque tous ses substrats devaient préalablement être ubiquitinylés. On a aussi longtemps considéré que l’ubiquitinylation et la dégradation étaient deux mécanismes découplés, et que le recrutement des conjugués ubiquitine s’effectuait directement par des sous-unités spécialisées du protéasome. La littérature récente remet en cause cette vue simplifiée. Elle suggère ainsi que la fraction des protéines hydrolysées par le protéasome, indépendamment de toute ubiquitinylation, a largement été sous-estimée, et que la reconnaissance des protéines ubiquitinylées fait intervenir des systèmes d’adressage complexes. Par ailleurs, elle indique un ordre d’organisation supérieur pour la voie ubiquitine/protéasome, une fraction du protéasome et des enzyme d’ubiquitinylation étant engagée dans des complexes supramoléculaires. Enfin, la dégradation protéasomique est altérée dans de nombreuses situations pathologiques. Elle constitue donc une cible thérapeutique dont les premières applications commencent à émerger.
Summary
The proteasome is the main intracellular proteolytic machinery. It is involved in all major cellular functions and decisions. It has long been thought that prior ubiquitinylation of almost all of its substrates was necessary for degradation. It has also long been considered that ubiquitinylation and degradation were two uncoupled mechanisms and that the recruitment of ubiquitinylated species was only performed by specialized subunits of the proteasome. The recent literature questions this simplified view. It also suggests that, on the one hand, the fraction of proteins hydrolyzed by the proteasome independently of their ubiquitinylation has largely been underestimated and, on the other hand, that the recognition of ubiquitinylated proteins involves complex addressing systems. Furthermore, it indicates a higher order structuration of the ubiquitin/proteasome pathway, a fraction of the proteasome and of ubiquitinylation enzymes being engaged in supramolecular complexes. Finally, proteasomal degradation is altered in a number of pathological situations. It, thus, constitutes a therapeutic target and the first applications are emerging.
Article body
Le protéasome 20S est une protéinase multimérique possédant une chambre protéolytique interne autocompartimentée qui s’associe à des complexes régulateurs pour former des structures comme le protéasome 26S (Figure 1A et 1B). Retrouvé dans le cytoplasme et le noyau, il constitue la principale machinerie protéolytique intracellulaire. Il est responsable de l’élimination des protéines anormales, âgées ou en excès, dont l’accumulation serait toxique. Il assure aussi la production des peptides antigéniques présentés par les molécules du CMH de classe I, la destruction contrôlée spatio-temporellement de nombreux régulateurs cellulaires ainsi que la maturation de différents précurseurs polypeptidiques. De ce fait, il est impliqué dans toutes les décisions cellulaires majeures comme la division, la différenciation, l’apoptose, l’intégration des signaux externes et la réponse immunitaire adaptative [1].
Figure 1
Protéasomes et voie ubiquitine.
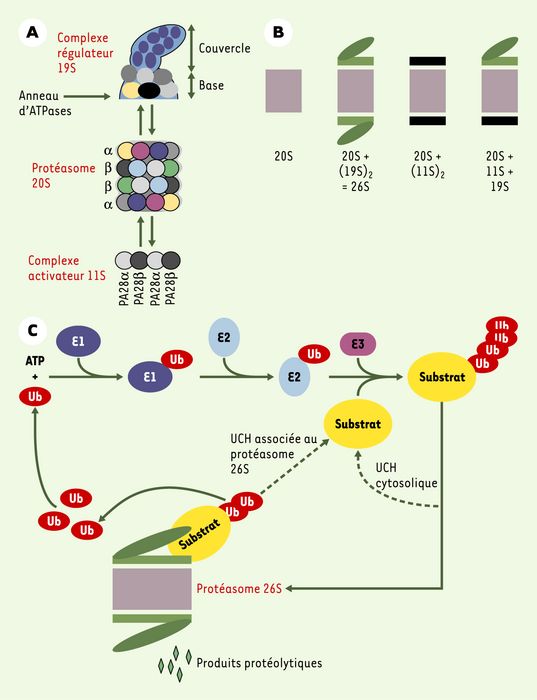
A. Le protéasome 20S et ses complexes régulateurs. Le protéasome 20S est le coeur catalytique de la machinerie protéasomique. Il s’agit d’un cylindre creux formé de 4 anneaux : 2 anneaux identiques de 7 sous-unités de type α et 2 autres, également identiques entre eux, de 7 sous-unités de type β. Les activités protéolytiques sont situées à l’intérieur de la structure et sont portées par 3 des 7 sous-unités β. Les anneaux α assurent l’interaction avec les complexes régulateurs du protéasome 20S et permettent l’entrée des substrats dans la chambre catalytique grâce à leur orifice central. Le complexe activateur 19S participe à la dégradation des protéines ubiquitinylées grâce à sa capacité à reconnaître les chaînes d’ubiquitine. Il possède un anneau d’ATPases chaperons participant à la déstructuration des substrats et, peut-être, à leur injection dans la cavité protéolytique, ainsi qu’une activité de désubiquitinylation (activité UCH, ubiquitin C-terminal hydrolase). Le complexe régulateur 11S est un anneau de 6 à 7 protéines de deux types différents : PA28α (proteasome activator) et PA28β. Il modifie les propriétés catalytiques du 20S, mais n’est pas capable de reconnaître des protéines ubiquitinylées. B. Les différents types de protéasome. Le protéasome 20S, retrouvé en partie libre dans la cellule, peut également s’associer avec des complexes 19S et 11S à chacune de ses extrémités. La particule traditionnellement appelée protéasome 26S est composé d’un 20S et de deux 19S. Les différentes particules sont présentes en quantités comparables dans les cellules mammifères. C. La voie ubiquitine/protéasome. L’ubiquitinylation des protéines fait intervenir trois types d’enzyme. E1 (ubiquitin-activating enzyme) est unique. Elle active l’ubiquitine et la transfère sur des enzymes E2 (ubiquitin-conjugating enzymes), qui sont au nombre d’une trentaine chez les mammifères. Directement, ou avec l’aide de composants E3, elles transfèrent et polymérisent l’ubiquitine sur les substrats. Dans la majorité des cas, les facteurs E3, dont certains sont multimériques, forment un pont moléculaire entre E2 et le substrat. D’autres transfèrent eux-mêmes l’ubiquitine sur les protéines acceptrices. Les E3 assurent l’essentiel de la spécificité de la réaction grâce à leur grand nombre (plusieurs centaines de E3 différentes dans une cellule eucaryote), bien qu’il existe une certaine redondance dans le système. Ainsi, une même protéine peut être reconnue par différentes E3 et une même E3 peut reconnaître différents substrats. L’ubiquitinylation est réglée par la signalisation intracellulaire via des modifications post-traductionnelles des substrats (phosphorylations, hydroxylation…) ou au travers de la modulation de l’activité des E3. Les protéines ubiquitinylées peuvent être désubiquitinylées par des UCH cytosoliques et recyclées. Dans le cas contraire, elles sont reconnues par des protéasomes contenant un complexe régulateur 19S qui assure la désubiquitinylation, la déstructuration et l’acheminement vers la cavité protéolytique. Si elles sont insuffisamment ou improprement ubiquitinylées, les protéines peuvent être recyclées grâce à une activité UCH portée par le complexe 19S lui-même. L’ubiquitine, qui est très stable, est réutilisée.
Il a longtemps été considéré que la dégradation protéasomique fonctionnait en deux temps disjoints. Dans une première phase, la plupart des substrats, sinon tous, devaient subir une modification post-traductionnelle particulière, la poly-ubiquitinylation. Celle-ci consiste en la conjugaison covalente de chaînes d’ubiquitine, une petite protéine conservée, ubiquitaire et abondante de 76 acides aminés (Figure 2). Cette importante découverte a d’ailleurs justifié l’attribution du prix Nobel 2004 de Chimie à Aaron Ciechanover, Avram Herschko et Irwin Rose ((→) m/s 2004, n° 12, p. 1161).
Figure 2
Les différentes chaînes d’ubiquitine.

A. La liaison isopeptidique. L’ubiquitine est conjuguée sur ses substrats via une liaison isopeptique entre son extrémité carboxyterminale et le groupement ε-NH2 des lysines acceptrices de ces derniers. Il s’agit d’une liaison amide, comme la liaison peptidique qui relie les acides aminés d’une même protéine entre eux. Dans certains cas, rares jusqu’à présent, l’ubiquitine peut se conjuger via une liaison peptidique classique entre son extrémité carboxyterminale et le NH2 terminal de la protéine acceptrice. B. Les chaînes d’ubiquitine. Les 7 lysines permettent la conjugaison de l’ubiquitine sur elle-même in vivo. Le rôle biologique des différentes chaînes n’est pas encore établi. Les chaînes K48 sont les plus mises en évidence, car elles servent à la dégradation des protéines. Elles sont toutefois également impliquées dans la régulation de l’activité de facteurs de transcription. L’existence de chaînes comportant des liaisons mixtes reste à caractériser. La longueur des chaînes est probablement un paramètre important pour leur action. Par exemple, des dimères d’ubiquitine couplés via la lysine 63 règlent l’endocytose de certains récepteurs membranaires, tandis que des chaînes plus longues modulent des activités kinases. C. Reconnaissance des substrats ubiquitinylés par le protéasome. Les substrats ubiquitinylés peuvent interagir directement ou indirectement avec le protéasome. Les chaînes d’ubiquitine peuvent ainsi reconnaître les sous-unités S5a et S6’ du complexe régulateur 19S. S6’ est l’une des ATPases de l’anneau de chaperons au contact du 20S. S5a occupe, quant à elle, une position intermédiaire entre la base et le couvercle du 19S. Cette protéine est reconnnue par les protéines navettes à UBD (domaine ubiquitin-like) et UBA (domaine se liant à l’ubiquitine) comme Rad23. L’ensemble des interactions des protéines à domaines UBD et UBA avec le protéasome 26S doit encore être précisé. CHIP : carboxy terminus of Hsp70-interacting protein ; Bag1 : Bcl2-associated athanogene.
Ensuite, les protéines ubiquitinylées étaient reconnues par des sous-unités spécifiques du protéasome 26S, puis désubiquitinylées, déstructurées et, enfin, hydrolysées. Cette vue s’est largement complexifiée ces dernières années. Il apparaît ainsi qu’une fraction des protéines, largement sous-estimée au départ, ne nécessite pas d’être ubiquitinylée pour être détruite par le protéasome [2-4]. Pour ce qui concerne les protéines ubiquitinylées, il est de plus en plus clair qu’elles font intervenir des systèmes d’adressage parfois complexes. Par ailleurs, le protéasome n’est pas forcément une particule libre dans la cellule. Il peut être engagé dans des supercomplexes comprenant aussi des enzymes d’ubiquitinylation, des chaperons moléculaires et d’autres protéines ou structures nécessaires à la dégradation de substrats particuliers. Ajoutons à cela que, suivant les conditions, la même protéine peut être reconnue via des mécanismes différents. Un premier exemple est celui de la protéine oncosuppressive p53 qui peut être ubiquitinylée par différentes enzymes mais, parfois, être aussi reconnue par le protéasome indépendamment de toute ubiquitinylation [5].
L’ubiquitinylation des protéines a déjà été décrite dans ces colonnes [6, 7]. Notre vue des mécanismes impliqués n’a pas changé substantiellement depuis. Ces derniers (Figure 1C) ne seront donc pas redétaillés ici. Pour plus d’information, le lecteur pourra se reporter à des revues exhaustives [1, 8]. Nous ne considérons ci-dessous que les points qui ont le plus modifié notre perception de la dégradation protéasomique dans le passé le plus récent.
L’ubiquitine
Les différents types de chaînes d’ubiquitine
L’ubiquitine est conjuguée à ses substrats via une liaison isopeptidique (Figure 2A) entre son extrémité carboxyterminale et le ε-NH2 de lysines acceptrices. Elle possède, elle-même, 7 lysines (K6, -11, -27, -29, -33, -48, -63) qui, chacune, permettent son autopolymérisation (Figure 2B). Les différents types de branchement confèrent une très grande richesse topologique aux chaînes d’ubiquitine, ce qui, vraisemblablement, permet l’adaptation à une grande variété de structures interactives. De fait, les différentes chaînes ont été impliquées dans des processus divers, non nécessairement liés à la destruction protéique [9-11]. Les plus représentées dans la cellule sont, de loin, les chaînes K48. Ce sont essentiellement elles qui sont impliquées dans le marquage des protéines à dégrader. Les chaînes K29, et peut-être K63, y contribuent probablement aussi, mais à un degré moindre. Soulignant la complexité du système, notons que les chaînes K48 peuvent aussi régler des fonctions non protéolytiques. Par ailleurs, comme nous le verrons plus loin, des chaînes non-K48 peuvent contrôler indirectement la reconnaissance des substrats à dégrader par le protéasome. Un enjeu majeur est de comprendre, au niveau moléculaire, ce qui détermine la nature des chaînes d’ubiquitine polymérisées sur les différents substrats et les interactions des différents types de chaînes avec les protéines cellulaires effectrices.
L’ubiquitine : au centre d’un réseau d’interactions protéines/protéines
Une notion récente importante est que l’ubiquitine, comme d’autres motifs structurellement reliés, sont des pièces essentielles dans un réseau complexe et dynamique d’interactions protéines/protéines au sein de la cellule. Du fait de l’implication de l’ubiquitine dans de nombreuses voies de signalisation [1, 9-11], ce « maillage » moléculaire ne concerne pas seulement la dégradation des protéines. Deux types de domaines se liant à l’ubiquitine, UBA (ubiquitin-associated domain) et UIM (ubiquitin-interacting motif), et deux autres, ubiquitin-like, UBD (ubiquitin domain) et UBX [12], sont retrouvés dans de nombreux polypeptides (Tableau I). De façon intéressante, une même protéine peut posséder deux types de motifs différents, éventuellement en exemplaires multiples. Même s’ils présentent des homologies de structure importantes, les différents domaines UBA et UIM n’ont pas la même affinité pour la mono- et la poly-ubiquitine. De plus, ils ne sont pas obligatoirement fonctionnellement équivalents, y compris au sein du même polypeptide. Si la base structurale de l’interaction entre UBA ou UIM, d’un côté, et ubiquitine, UBD ou UBX, de l’autre, commence à s’éclaircir, les mécanismes responsables de la dynamique des associations sont encore loin d’être compris.
Tableau I
Domaines se liant à l’ubiquitine et domaines ubiquitin-like.
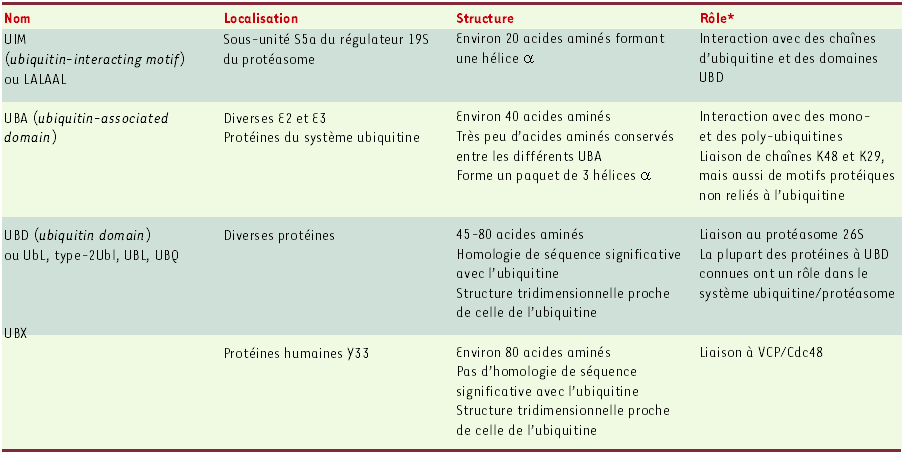
* Les rôles indiqués n’excluent pas que les motifs décrits puissent avoir d’autres rôles non encore identifiés. VCP : valosin-containing protein (homologue du régulateur du cycle cellulaire Cdc48).
Dégradation protéasomique des substrats ubiquitinylés
Reconnaissance directe ou adressage indirect des substrats ubiquitinylés au protéasome ?
De multiples mécanismes sont responsables de l’adressage des substrats au protéasome, même s’il n’existe pas encore d’accord général sur leur identité et leur nombre exact [12-14]. Cette variété reflète probablement la diversité des situations à laquelle les cellules doivent faire face. On peut, par exemple, facilement comprendre que la dégradation d’une protéine qui doit être extraite du réticulum endoplasmique pour être détruite n’obéisse pas aux mêmes contraintes que celle d’une protéine nucléaire.
Il est probable qu’une fraction des protéines ubiquitinylées soit reconnue directement par le protéasome. Ainsi, deux sous-unités du complexe régulateur 19S du protéasome (Figure 2C), S6’ et S5a, peuvent se lier à des chaînes d’ubiquitine K48, pour peu que celles-ci soient suffisamment longues [15-17]. Au niveau de S5a, l’interaction fait intervenir des motifs UIM. Les positions spatiales de S6’ et de S5a sont intéressantes à considérer. En effet, la première fait partie de l’anneau d’ATPases chaperons de la base du complexe régulateur 19S au contact direct du protéasome 20S, tandis que la seconde interagit avec cet anneau (Figure 2C). Cette organisation topologique favorise probablement la déstructuration des substrats et leur injection dans la chambre catalytique du 20S après désubiquitinylation par une autre sous-unité du complexe 19S, Rpn11 [18, 19].
En fait, il est possible (bien que la démonstration formelle soit encore manquante) que la plupart des protéines ubiquitinylées soient adressées au protéasome grâce à des protéines navettes se liant d’un côté à des chaînes d’ubiquitine et, de l’autre, au complexe régulateur 19S [12-14]. Chez la levure, l’équivalent de S5a, dont une partie est soluble, semble capable d’assurer cette fonction, la liaison des chaînes d’ubiquitine à S5a étant assurée par un motif UIM. Chez les mammifères, il se pourrait que ce mécanisme n’opère pas, S5a n’ayant pas encore été visualisée sous forme libre. Parmi les navettes moléculaires non protéasomiques, les protéines de la famille Rad23 (hHR23α et -β chez l’homme), initialement caractérisées pour leur implication dans la réparation de l’ADN, sont les mieux étudiées [12-14]. Elles possèdent un domaine UBD et deux domaines UBA. Ces derniers se lient aux chaînes d’ubiquitine, tandis que le domaine UBD interagit avec l’un des deux domaines UIM de S5a (Figure 2C). Si l’interaction avec l’UIM de S5a représente bien un premier point d’ancrage au protéasome, les données expérimentales suggèrent qu’il en existe au moins un autre dans le complexe régulateur 19S.
Association du protéasome avec les enzymes d’ubiquitinylation
Comme cela est expliqué dans la Figure 1C, le transfert de l’ubiquitine sur les substrats est assuré par deux classes d’enzymes spécialisées, E2 et E3 [1, 6, 7]. Des approches biochimiques, protéomiques et de criblage double hybride dans la levure ont permis de mettre en évidence l’inclusion du protéasome dans des supercomplexes contenant différentes E2 et E3. Cela a été observé chez la levure, les plantes comme Arabidopsis thaliana, les métazoaires inférieurs comme Caenorhabditis elegans ou les cellules de mammifères (Figure 3A). En fonction des situations, l’interaction est directe ou indirecte. Au moins dans certains cas, des protéines à UBD et UBA pourraient jouer le rôle de ponts moléculaires. Il semble en être ainsi de la protéine hPLIC-2 (ubiquitin-like product chap1/Dsk2). Pour associer la E3 E6-AP (E6- associated protein) au protéasome, elle se lierait à S5a via son domaine UBA, ainsi qu’à un autre site du complexe régulateur 19S via son domaine UBD [20]. Notons ici que hPLIC-2 pourrait ponter d’autres E3 au protéasome, d’une part, et que tous les UBA ne sont pas capables de se lier à S5a, d’autre part [20, 21].
Figure 3
Engagement du protéasome dans des complexes supramoléculaires.
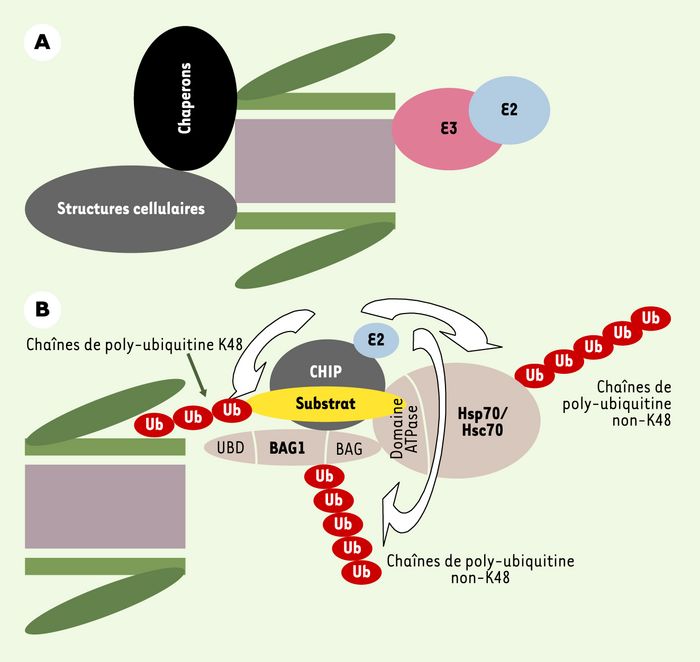
A. Interactions du protéasome avec d’autres composants de la cellule. Le protéasome peut interagir avec des enzymes d’ubiquitinylation et des chaperons moléculaires, mais aussi avec diverses structures cellulaires comme les systèmes de translocation qui permettent d’extraire les protéines du réticulum endoplasmique. B. Interactions complexes entre le protéasome, des chaperons moléculaires, des enzymes d’ubiquitinylation et des molécules adaptatrices. Cet exemple montre comment des interactions moléculaires complexes peuvent stimuler la dégradation de substrats ubiquitinylés par le protéasome. CHIP (carboxy terminus of Hsp70-interacting protein) est un cochaperon qui interagit avec les protéines chaperons de la famille Hsp70/Hsc70 (protéines de choc thermique inductible/constitutive). Elle possède une activité E3 permettant la formation de différents types de chaînes de poly-ubiquitine. En interagissant avec le domaine ATPase de Hsp70/Hsc70, elle inhibe le recyclage de substrats par ces chaperons. De façon concomitante, elle stimule l’ubiquitinylation par des chaînes K48 des protéines à dégrader par une E2 qui lui est associée. Par ailleurs, elle interagit avec une protéine adaptatrice, Bag1 (Bcl2-associated athanogene), qui se lie au complexe régulateur 19S grâce à un domaine UBD. Cette interaction, qui est capitale pour le maintien du supercomplexe, favorise aussi le transfert du substrat ubiquitinylé au protéasome via la conjugaison de chaînes non-K48 sur Bag1. Parallèment, CHIP stimule également la formation de chaînes non-K48 sur Hsp70/Hsc70. Ni la manière dont l’ubiquitinylation de Bag1 stimule la dégradation des protéines substrats, ni la raison de l’ubiquitinylation de Hsp70/Hsc70 ne sont élucidées.
Une interaction directe E3/protéasome particulère est intéressante à considérer ici. La parkine E3, qui est le produit du gène responsable de la maladie de Parkinson juvénile récessive, interagirait directement avec S5a via son domaine UBD. La mutation R42P trouvée chez certains patients induit un changement de conformation dans le domaine UBD, ce qui diminuerait l’interaction avec le protéasome et constituerait la cause structurale/biochimique de la maladie de Parkinson chez ces patients [22]. Pour terminer, mentionnons que des enzymes de désubiquitinylation comme l’isopeptidase Ubp6 (ubiquitin-specific protease 6) peuvent aussi s’associer avec le protéasome, sans que la raison moléculaire de cette association n’ait encore été élucidée [23, 24].
Implications des chaperons moléculaires dans l’adressage au protéasome
Les chaperons moléculaires sont bien connus comme machines de remise en conformation des protéines agrégées ou dénaturées. Très tôt, ils ont été suspectés d’intervenir dans le contrôle de la dégradation de substrats divers [1, 25]. De fait, différents chaperons s’associent avec le protéasome (Figure 3A) [13]. Des arguments fonctionnels existent pour impliquer les chaperons Hsc70/Hsp70 (protéines de choc thermique constitutive/inductible), Hsp90 et VCP/Cdc48 (valosin-containing protein, homologue du régulateur du cycle cellulaire Cdc48) dans la dégradation de substrats tels que des récepteurs membranaires, des protéines cytosoliques, des facteurs de transcription… [13]. Une des idées prévalant actuellement pour VCP/Cdc48 est qu’elle interviendrait pour extraire des protéines ubiquitinylées de complexes multimériques de manière à faciliter leur reconnaissance par le protéasome. En ce qui concerne Hsp90, elle fait intervenir d’autres molécules comme son cochaperon CHIP, qui est une E3, et des molécules adaptatrices à domaine UBD, comme Bag-1 (voirFigure 3B et [13], pour exemples).
Dégradation protéasomique des protéines non ubiquitinylées
Les protéines ne nécessitant pas d’ubiquitinylation pour être dégradées par le protéasome ont, jusqu’à récemment, été considérées comme de rares exceptions. L’accumulation actuelle de nouveaux exemples (Tableau II) [2-4] suggère que cette catégorie de substrats a probablement été largement sous-estimée dans le passé. Renforçant cette idée, les banques de données indiquent qu’environ 1 % des protéines mammifères sont dépourvues de lysines. Il a néanmoins été proposé que ces mêmes protéines pourraient être ubiquitinylées sur leur extrémité aminoterminale via la formation d’une liaison peptidique.
Tableau II
Protéines dégradées sans ubiquitinylation préalable.
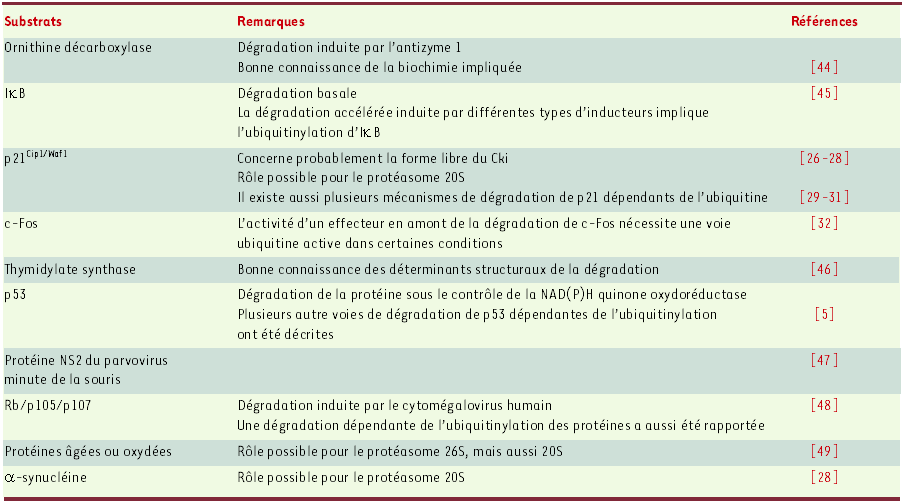
Seuls des substrats faisant l’objet, in vivo, d’une dégradation protéasomique indépendante de leur ubiquitinylation propre sont mentionnés ici.
La protéolyse doit être considérée avec le fait que n’importe quelle protéine dans la cellule constitue une population de molécules hétérogènes au niveau de la fonction, de la localisation, du partenariat et des modifications post-traductionnelles. Il s’ensuit qu’elles peuvent subir divers modes de dégradation en fonction de ces paramètres et des conditions physiologiques. Par exemple, p21CIP1/WAF1, l’inhibiteur de kinase dépendante des cyclines - qui a été l’une des premières protéines identifiées pour être dégradée indépendamment de son ubiquitinylation [26-28] - peut être déstabilisée par ubiquitinylation dans certaines conditions [29-31]. Par ailleurs, si le substrat n’est pas ubiquitinylé lui-même, il est possible, dans certains cas, que des chaînes d’ubiquitine soient apportées en trans par un partenaire d’interaction. Enfin, si l’ubiquitine n’est pas requise dans les étapes finales de la reconnaissance de certains substrats par le protéasome, elle peut indirectement régler la dégradation d’effecteurs en amont de leur destruction, comme cela pourrait être le cas pour la proto-oncoprotéine c-Fos, au moins dans certaines situations [32].
Deux questions importantes se posent. La première est de savoir quelles sont la (ou les) forme(s) de protéasome impliquée(s) dans la dégradation des protéines non ubiquitinylées. La seconde est de déterminer s’il existe des systèmes d’adressage spécifiques, comme il en existe pour les substrats ubiquitinylés. Pour le second point, aucune information n’est disponible à l’heure actuelle. Pour le premier, en revanche, il semble que plusieurs formes de protéasome puissent participer à la destruction indépendante de l’ubiquitine. Ainsi, des expériences réalisées in vitro ont montré depuis longtemps que le protéasome 26S peut dégrader l’ornithine décarboxylase. Cependant, des particules hybrides 20S + 19S + 11S semblent plus actives [33]. Le point récent le plus surprenant est la suggestion selon laquelle le protéasome 20S seul pourrait aussi être un acteur majeur de la dégradation indépendante de l’ubiquitine [28]. Bien que ce soit la particule la plus abondante dans certaines cellules mammifères, on a longtemps pensé qu’il s’agissait simplement d’une forme latente, en attente d’interaction avec des complexes activateurs. Cette supposition reposait sur des analyses structurales montrant que les deux orifices apicaux du protéasome étaient obturés par les extrémités flexibles des sous-unités α en l’absence de complexes régulateurs [1]. En fait, des travaux récents montrent que certaines protéines faiblement structurées, comme p21 ou l’α-synucléine [28], sont capables de désorganiser cette obturation et de trouver, seules, leur chemin vers la chambre catalytique. Une question majeure à résoudre maintenant est l’identification des substrats cibles des différents types de complexes protéasomiques in vivo.
Perturbations du système ubiquitine/protéasome et perspectives thérapeutiques
Faisant partie intégrante des cascades de signalisation intracellulaire, il n’est pas étonnant que la dégradation protéasomique soit altérée dans de nombreuses situations pathologiques. Les mécanismes mis en évidence n’impliquent pour le moment que l’ubiquitinylation de régulateurs cellulaires clés. Le domaine évoluant rapidement, il est vraisemblable que des anomalies d’adressage au protéasome seront aussi découvertes dans un futur proche.
Les maladies étudiées incluent, entre autres, des neurodégénérescences, des maladies génétiques, des déficits immunitaires, des désordres hématopoïétiques et les cancers [1, 34, 35]. Dans ce dernier cas, il est intéressant de noter que certains des composants des systèmes d’ubiquitinylation sont soit des oncogènes (par exemple, Mdm2, mouse double minute 2), soit des suppresseurs de tumeurs (par exemple, pVHL, von Hippel-Lindau disease tumor suppressor) [36]. Soulignons, dans le cas des cancers, que les effets transformants ne concernent pas seulement la division cellulaire [37]. D’autres processus peuvent être atteints : on peut citer des anomalies de différenciation ou l’acquisition de la résistance aux agents génotoxiques. Les cellules peuvent également acquérir la capacité de survivre dans des conditions acides ou hypoxiques, de manifester une activité invasive ou métastatique, de stimuler l’angiogenèse ou d’échapper au système immunitaire. Par ailleurs, certains virus tirent aussi profit du système ubiquitine/protéasome (Ub/Pr) pour exercer leurs effets délétères, se protéger des réponses interféron antivirale ou immune adaptatrice et asservir les cellules infectées à leurs besoins [38, 39].
Toutes ces observations font du système Ub/Pr une cible attractive pour le développement de nouveaux outils diagnostiques et thérapeutiques [1, 34, 35]. Trois axes de travaux soulignent déjà la pertinence de cette dernière réflexion. La première est qu’un inhibiteur du protéasome (Velcade®), plus toxique pour beaucoup de cellules cancéreuses que pour les cellules non transformées, a déjà montré une efficacité importante dans le traitement du myélome multiple [40]. Il est actuellement en essai clinique de phase II pour d’autres cancers, bien que les conséquences moléculaires de son action ne soient pas encore précisément comprises. La seconde est que l’action de l’acide rétinoïque et du trioxyde d’arsenic, utilisés dans le traitement de la leucémie myéloïde chronique, s’explique, au moins en partie, par la déstabilisation de la protéine oncogénique PML-RARα (pro-myelocytic leukemia/retinoic acid receptor-α) produite par translocation chromosomique [41]. Enfin, le 17-allylaminogeldanamycine, un inhibiteur du chaperon Hsp90, impliqué - comme nous l’avons déjà mentionné - dans le contrôle de la stabilité de nombreuses protéines, est en cours d’essais cliniques anticancer. Il pourrait aussi trouver des applications dans le cadre de maladies virales, neurologiques et auto-immunes [42, 43]. Gageons que ceci ne soit qu’un début à de nombreuses applications futures.
Conclusions
En résumé, on peut affirmer que notre perception du système ubiquitine/protéasome a fortement évolué ces dernières années. Les points les plus saillants concernent certainement l’inclusion des enzymes d’ubiquitinylation et du protéasome dans des complexes supramoléculaires, ainsi que la mise en évidence de processus complexes d’adressage des protéines à dégrader à la machinerie protéolytique. Caractériser les différents composants impliqués, comprendre leur dynamique, relier leur(s) activité(s) à la signalisation et à la compartimentalisation intracellulaire constituent aujourd’hui des enjeux majeurs du domaine. De façon intéressante, les premières substances affectant la dégradation des protéines ont commencé à montrer des effets bénéfiques dans le traitement de certains cancers. Considérant l’engagement de plus en plus fort de l’industrie dans cette recherche, on peut raisonnablement espérer de nombreuses applications futures. Par ailleurs, il doit être souligné que la dissection du système ubiquitine/protéasome - au-delà du fait de permettre de mieux comprendre la protéolyse intracellulaire - a ouvert de nombreux autres champs disciplinaires : ces dernières années ont notamment montré que l’ubiquitine ne constitue pas un modificateur post-traductionnel peptidique unique, mais représente seulement le chef de file d’une famille de telles molécules réglant des mécanismes moléculaires et cellulaires très diversifiés. Il est aussi apparu que la nature modulaire du protéasome 26S permettait à certains de ses composants d’intervenir dans des fonctions non protéolytiques, comme la réplication de l’ADN ou la transcription. Souhaitons que les investigations à ces deux derniers niveaux soient aussi riches en développements fondamentaux qu’en termes de compréhension de nombreuses maladies et de leur traitement.
Appendices
Remerciements
Nos travaux ont été soutenus par l’ARC, la Ligue contre le cancer, HFSP, le programme Quality of Life de la Commission européenne, le CNRS et le programme ACI du Ministère de la Recherche.
Références
- 1. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway : destruction for the sake of construction. Physiol Rev 2002 ; 82 : 373-428.
- 2. Forster A, Hill CP. Proteasome degradation : enter the substrate. Trends Cell Biol 2003 ; 13 : 550-3.
- 3. Orlowski M, Wilk S. Ubiquitin-independent proteolytic functions of the proteasome. Arch Biochem Biophys 2003 ; 415 : 1-5.
- 4. Verma R, Deshaies RJ. A proteasome howdunit : the case of the missing signal. Cell 2000 ; 101 : 341-4.
- 5. Asher G, Lotem J, Sachs L, et al. Mdm-2 and ubiquitin-independent p53 proteasomal degradation regulated by NQO1. Proc Natl Acad Sci USA 2002 ; 99 : 13125-30.
- 6. Ferrara P, Acquaviva C, Bossis G, et al. Protéolyse intracellulaire et tumorigenèse. Med Sci (Paris) 2001 ; 17 : 5-13.
- 7. Coux O, Piechaczyk M. Le système ubiquitine/protéasome : un ensemble (de) complexe(s) pour dégrader les protéines. Med Sci (Paris) 2000 ; 16 : 623-9.
- 8. Coux O. The 26S proteasome. Prog Mol Subcell Biol 2002 ; 29 : 85-107.
- 9. Sun L, Chen ZJ. The novel functions of ubiquitination in signaling. Curr Opin Cell Biol 2004 ; 16 : 119-26.
- 10. Pickart CM. Ubiquitin enters the new millennium. Mol Cell 2001 ; 8 : 499-504.
- 11. Weissman AM. Themes and variations on ubiquitynation. Nat Rev Mol Cell Biol 2001 ; 2 : 169-78.
- 12. Buchberger A. From UBA to UBX : new words in the ubiquitin vocabulary. TrendsCell Biol 2002 ; 12 : 216-21.
- 13. Farras R, Bossis G, Andermarcher E, et al. Mechanisms of delivery of ubiquitinylated proteins to the proteasome : new targets for anti-cancer therapy. Crit Rev Oncol Hematol 2005 (sous presse).
- 14. Hartmann-Petersen R, Seeger M, Gordon C. Transferring substrates to the 26S proteasome. Trends Biochem Sci 2003 ; 28 : 26-31.
- 15. Deveraux Q, Ustrell V, Pickart C, Rechsteiner M. A 26 S protease subunit that binds ubiquitin conjugates. J Biol Chem 1994 ; 269 : 7059-61.
- 16. Lam YA, Lawson TG, Velayutham M, et al. A proteasomal ATPase subunit recognizes the polyubiquitin degradation signal. Nature 2002 ; 416 : 763-7.
- 17. Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J 2000 ; 19 : 94-102.
- 18. Yao T, Cohen RE. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature 2002 ; 419 : 403-7.
- 19. Verma R, Aravind L, Oania R, et al. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 2002 ; 298 : 611-5.
- 20. Kleijnen MF, Shih AH, Zhou P, et al. The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol Cell 2000 ; 6 : 409-19.
- 21. Kleijnen MF, Alarcon RM, Howley PM. The ubiquitin-associated domain of hPLIC-2 interacts with the proteasome. Mol Biol Cell 2003 ; 14 : 3868-75.
- 22. Sakata E, Yamaguchi Y, Kurimoto E, et al. Parkin binds the Rpn10 subunit of 26S proteasomes through its ubiquitin-like domain. EMBO Rep 2003 ; 4 : 301-6.
- 23. Verma R, Chen S, Feldman R, et al. Proteasomal proteomics : identification of nucleotide-sensitive proteasome-interacting proteins by mass spectrometric analysis of affinity-purified proteasomes. Mol Biol Cell 2000 ; 11 : 3425-39.
- 24. Leggett DS, Hanna J, Borodovsky A, et al. Multiple associated proteins regulate proteasome structure and function. Mol Cell 2002 ; 10 : 495-507.
- 25. McClellan AJ, Frydman J. Molecular chaperones and the art of recognizing a lost cause. Nat Cell Biol 2001 ; 3 : E51-3.
- 26. Sheaff RJ, Singer JD, Swanger J et al. Proteasomal turnover of p21Cip1 does not require p21Cip1 ubiquitination. Mol Cell 2000 ; 5 : 403-10.
- 27. Touitou R, Richardson J, Bose S, et al. A degradation signal located in the C-terminus of p21WAF1/CIP1 is a binding site for the C8 alpha-subunit of the 20S proteasome. EMBO J 2001 ; 20 : 2367-75.
- 28. Liu CW, Corboy MJ, DeMartino GN, Thomas PJ. Endoproteolytic activity of the proteasome. Science 2003 ; 299 : 408-11.
- 29. Bloom J, Amador V, Bartolini F, et al. Proteasome-mediated degradation of p21 via N-terminal ubiquitinylation. Cell 2003 ; 115 : 71-82.
- 30. Bornstein G, Bloom J, Sitry-Shevah D, et al. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J Biol Chem 2003 ; 278 : 25752-7.
- 31. Bendjennat M, Boulaire J, Jascur T, et al. UV irradiation triggers ubiquitin-dependent degradation of p21(WAF1) to promote DNA repair. Cell 2003 ; 114 : 599-610.
- 32. Bossis G, Ferrara P, Acquaviva C, et al. c-Fos proto-oncoprotein is degraded by the proteasome independently of its own ubiquitinylation in vivo. Mol Cell Biol 2003 ; 23 : 7425-36.
- 33. Tanahashi N, Murakami Y, Minami Y, et al. Hybrid proteasomes. Induction by interferon-gamma and contribution to ATP-dependent proteolysis. J Biol Chem 2000 ; 275 : 14336-45.
- 34. Ciechanover A, Schwartz AL. Ubiquitin-mediated degradation of cellular proteins in health and disease. Hepatology 2002 ; 35 : 3-6.
- 35. Plemper RK, Hammond AL. Protein degradation in human disease. Prog Mol Subcell Biol 2002 ; 29 : 61-84.
- 36. Pagano M, Benmaamar R. When protein destruction runs amok, malignancy is on the loose. Cancer Cell 2003 ; 4 : 251-6.
- 37. Sherr CJ. Principles of tumor suppression. Cell 2004 ; 116 : 235-46.
- 38. Liu YC. Ubiquitin ligases and the immune response. Annu Rev Immunol 2004 ; 22 : 81-127.
- 39. Shackelford J, Pagano JS. Tumor viruses and cell signaling pathways : deubiquitination versus ubiquitination. Mol Cell Biol 2004 ; 24 : 5089-93.
- 40. Adams J. The proteasome : a suitable antineoplastic target. Nat Rev Cancer 2004 ; 4 : 349-60.
- 41. Jing Y. The PML-RARalpha fusion protein and targeted therapy for acute promyelocytic leukemia. Leuk Lymphoma 2004 ; 45 : 639-48.
- 42. Zhang H, Burrows F. Targeting multiple signal transduction pathways through inhibition of Hsp90. J Mol Med 2004 ; 82 : 488-99.
- 43. Kamal A, Boehm MF, Burrows FJ. Therapeutic and diagnostic implications of Hsp90 activation. Trends Mol Med 2004 ; 10 : 283-90.
- 44. Zhang M, Pickart CM, Coffino P. Determinants of proteasome recognition of ornithine decarboxylase, a ubiquitin-independent substrate. EMBO J 2003 ; 22 : 1488-96.
- 45. Krappmann D, Wulczyn FG, Scheidereit C. Different mechanisms control signal-induced degradation and basal turnover of the NF-kappaB inhibitor IkappaB alpha in vivo. EMBO J 1996 ; 15 : 6716-26.
- 46. Forsthoelfel AM, Pena MMO, Xing YY, et al. Structural determinants of the intracellular degradation of human thymidylate synthase. Biochemistry 2004 ; 43 : 1972-9.
- 47. Miller CL, Pintel DJ. The NS2 protein generated by the parvovirus minute virus of mice is degraded by the proteasome in a manner independent of ubiquitin chain elongation or activation. Virology 2001 ; 285 : 346-55.
- 48. Kalejta RF, Shenk T. Proteasome-dependent, ubiquitin-independent degradation of the Rb family of tumor suppressors by the human cytomegalovirus pp71 protein. Proc Natl Acad Sci USA 2003 ; 100 : 3263-8.
- 49. Shringarpure R, Grune T, Mehlhase J, Davies KJ. Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. J Biol Chem 2003 ; 278 : 311-8.
List of figures
Figure 1
Protéasomes et voie ubiquitine.
A. Le protéasome 20S et ses complexes régulateurs. Le protéasome 20S est le coeur catalytique de la machinerie protéasomique. Il s’agit d’un cylindre creux formé de 4 anneaux : 2 anneaux identiques de 7 sous-unités de type α et 2 autres, également identiques entre eux, de 7 sous-unités de type β. Les activités protéolytiques sont situées à l’intérieur de la structure et sont portées par 3 des 7 sous-unités β. Les anneaux α assurent l’interaction avec les complexes régulateurs du protéasome 20S et permettent l’entrée des substrats dans la chambre catalytique grâce à leur orifice central. Le complexe activateur 19S participe à la dégradation des protéines ubiquitinylées grâce à sa capacité à reconnaître les chaînes d’ubiquitine. Il possède un anneau d’ATPases chaperons participant à la déstructuration des substrats et, peut-être, à leur injection dans la cavité protéolytique, ainsi qu’une activité de désubiquitinylation (activité UCH, ubiquitin C-terminal hydrolase). Le complexe régulateur 11S est un anneau de 6 à 7 protéines de deux types différents : PA28α (proteasome activator) et PA28β. Il modifie les propriétés catalytiques du 20S, mais n’est pas capable de reconnaître des protéines ubiquitinylées. B. Les différents types de protéasome. Le protéasome 20S, retrouvé en partie libre dans la cellule, peut également s’associer avec des complexes 19S et 11S à chacune de ses extrémités. La particule traditionnellement appelée protéasome 26S est composé d’un 20S et de deux 19S. Les différentes particules sont présentes en quantités comparables dans les cellules mammifères. C. La voie ubiquitine/protéasome. L’ubiquitinylation des protéines fait intervenir trois types d’enzyme. E1 (ubiquitin-activating enzyme) est unique. Elle active l’ubiquitine et la transfère sur des enzymes E2 (ubiquitin-conjugating enzymes), qui sont au nombre d’une trentaine chez les mammifères. Directement, ou avec l’aide de composants E3, elles transfèrent et polymérisent l’ubiquitine sur les substrats. Dans la majorité des cas, les facteurs E3, dont certains sont multimériques, forment un pont moléculaire entre E2 et le substrat. D’autres transfèrent eux-mêmes l’ubiquitine sur les protéines acceptrices. Les E3 assurent l’essentiel de la spécificité de la réaction grâce à leur grand nombre (plusieurs centaines de E3 différentes dans une cellule eucaryote), bien qu’il existe une certaine redondance dans le système. Ainsi, une même protéine peut être reconnue par différentes E3 et une même E3 peut reconnaître différents substrats. L’ubiquitinylation est réglée par la signalisation intracellulaire via des modifications post-traductionnelles des substrats (phosphorylations, hydroxylation…) ou au travers de la modulation de l’activité des E3. Les protéines ubiquitinylées peuvent être désubiquitinylées par des UCH cytosoliques et recyclées. Dans le cas contraire, elles sont reconnues par des protéasomes contenant un complexe régulateur 19S qui assure la désubiquitinylation, la déstructuration et l’acheminement vers la cavité protéolytique. Si elles sont insuffisamment ou improprement ubiquitinylées, les protéines peuvent être recyclées grâce à une activité UCH portée par le complexe 19S lui-même. L’ubiquitine, qui est très stable, est réutilisée.
Figure 2
Les différentes chaînes d’ubiquitine.
A. La liaison isopeptidique. L’ubiquitine est conjuguée sur ses substrats via une liaison isopeptique entre son extrémité carboxyterminale et le groupement ε-NH2 des lysines acceptrices de ces derniers. Il s’agit d’une liaison amide, comme la liaison peptidique qui relie les acides aminés d’une même protéine entre eux. Dans certains cas, rares jusqu’à présent, l’ubiquitine peut se conjuger via une liaison peptidique classique entre son extrémité carboxyterminale et le NH2 terminal de la protéine acceptrice. B. Les chaînes d’ubiquitine. Les 7 lysines permettent la conjugaison de l’ubiquitine sur elle-même in vivo. Le rôle biologique des différentes chaînes n’est pas encore établi. Les chaînes K48 sont les plus mises en évidence, car elles servent à la dégradation des protéines. Elles sont toutefois également impliquées dans la régulation de l’activité de facteurs de transcription. L’existence de chaînes comportant des liaisons mixtes reste à caractériser. La longueur des chaînes est probablement un paramètre important pour leur action. Par exemple, des dimères d’ubiquitine couplés via la lysine 63 règlent l’endocytose de certains récepteurs membranaires, tandis que des chaînes plus longues modulent des activités kinases. C. Reconnaissance des substrats ubiquitinylés par le protéasome. Les substrats ubiquitinylés peuvent interagir directement ou indirectement avec le protéasome. Les chaînes d’ubiquitine peuvent ainsi reconnaître les sous-unités S5a et S6’ du complexe régulateur 19S. S6’ est l’une des ATPases de l’anneau de chaperons au contact du 20S. S5a occupe, quant à elle, une position intermédiaire entre la base et le couvercle du 19S. Cette protéine est reconnnue par les protéines navettes à UBD (domaine ubiquitin-like) et UBA (domaine se liant à l’ubiquitine) comme Rad23. L’ensemble des interactions des protéines à domaines UBD et UBA avec le protéasome 26S doit encore être précisé. CHIP : carboxy terminus of Hsp70-interacting protein ; Bag1 : Bcl2-associated athanogene.
Figure 3
Engagement du protéasome dans des complexes supramoléculaires.
A. Interactions du protéasome avec d’autres composants de la cellule. Le protéasome peut interagir avec des enzymes d’ubiquitinylation et des chaperons moléculaires, mais aussi avec diverses structures cellulaires comme les systèmes de translocation qui permettent d’extraire les protéines du réticulum endoplasmique. B. Interactions complexes entre le protéasome, des chaperons moléculaires, des enzymes d’ubiquitinylation et des molécules adaptatrices. Cet exemple montre comment des interactions moléculaires complexes peuvent stimuler la dégradation de substrats ubiquitinylés par le protéasome. CHIP (carboxy terminus of Hsp70-interacting protein) est un cochaperon qui interagit avec les protéines chaperons de la famille Hsp70/Hsc70 (protéines de choc thermique inductible/constitutive). Elle possède une activité E3 permettant la formation de différents types de chaînes de poly-ubiquitine. En interagissant avec le domaine ATPase de Hsp70/Hsc70, elle inhibe le recyclage de substrats par ces chaperons. De façon concomitante, elle stimule l’ubiquitinylation par des chaînes K48 des protéines à dégrader par une E2 qui lui est associée. Par ailleurs, elle interagit avec une protéine adaptatrice, Bag1 (Bcl2-associated athanogene), qui se lie au complexe régulateur 19S grâce à un domaine UBD. Cette interaction, qui est capitale pour le maintien du supercomplexe, favorise aussi le transfert du substrat ubiquitinylé au protéasome via la conjugaison de chaînes non-K48 sur Bag1. Parallèment, CHIP stimule également la formation de chaînes non-K48 sur Hsp70/Hsc70. Ni la manière dont l’ubiquitinylation de Bag1 stimule la dégradation des protéines substrats, ni la raison de l’ubiquitinylation de Hsp70/Hsc70 ne sont élucidées.
List of tables
Tableau I
Domaines se liant à l’ubiquitine et domaines ubiquitin-like.
* Les rôles indiqués n’excluent pas que les motifs décrits puissent avoir d’autres rôles non encore identifiés. VCP : valosin-containing protein (homologue du régulateur du cycle cellulaire Cdc48).
Tableau II
Protéines dégradées sans ubiquitinylation préalable.
Seuls des substrats faisant l’objet, in vivo, d’une dégradation protéasomique indépendante de leur ubiquitinylation propre sont mentionnés ici.