Abstracts
Résumé
Les retards mentaux liés au chromosome X (RMLX), qui touchent 1,8 garçons pour 1 000 naissances masculines, sont classiquement divisés en formes syndromiques et formes non spécifiques, selon la présence ou non de signes particuliers associés au retard mental. L’extrême hétérogénéité phénotypique et allélique, parfois visible au sein d’une même famille, complique toutefois cette classification. L’évaluation rétrospective appronfondie des familles atteintes, une fois la mutation identifiée dans un gène, devrait aider à clarifier la situation et faciliter la prise en charge du diagnostic moléculaire de ces retards mentaux. L’analyse des protéines produites par les 60 gènes de RMLX actuellement identifiés montre une grande diversité des fonctions biologiques affectées dans le retard mental. Dans cette revue, nous présenterons les données récentes concernant trois gènes, FMR1, ARX et le gène de l’oligophrénine 1, qui non seulement illustrent la complexité des RMLX, mais soulignent aussi l’importance des voies de signalisation impliquées dans la régulation de l’expression génique, ainsi que celles relayées pas les GTPases Rho dans la maturation et la plasticité neuronale.
Summary
X-linked mental retardation (XLMR) affects 1.8 ‰ male births and is usually categorized as “syndromic” (MRXS) or “non-specific” (MRX) forms according to the presence or absence of specific signs in addition to the MR. Up to 60 genes have been implicated in XLMR and certain mutations can alternatively lead to MRXS or MRX. Indeed the extreme phenotypic and allelic heterogeneity of XLMR makes the classification of most genes difficult. Therefore, following identification of new genes, accurate retrospective clinical evaluation of patients and their families is necessary to aid the molecular diagnosis and the classification of this heterogeneous group of disorders. Analyses of the protein products corresponding to XLMR genes show a great diversity of cellular pathways involved in MR. Common mechanisms are beginning to emerge : a first group of proteins belongs to the Rho and Rab GTPase signaling pathways involved in neuronal differentiation and synaptic plasticity and a second group is related to the regulation of gene expression. In this review, we illustrate the complexity of XLMR conditions and present recent data about the FMR1, ARX and Oligophrenin 1 genes.
Article body
Retards mentaux liés à l’X
La préférence masculine
Depuis des décennies, les pédopsychiatres savent que les retards mentaux ont une incidence plus élevée chez les garçons que chez les filles. La découverte du syndrome de l’X fragile, d’abord identifié par l’observation d’une cassure spontanée en Xq28, puis par la mise en évidence de son mécanisme pathogénique particulier (expansion du triplet CGG), sembla pendant un temps fournir l’explication de ce déséquilibre. Mais il est rapidement apparu qu’il existe de nombreux autres gènes, localisés sur le chromosome X, impliqués dans des retards mentaux liés à l’X chez le garçon (RMLX).
D’innombrables variétés
Le retard mental (RM) se définit par un quotient intellectuel < 70. Par rapport au nombre de naissances, on compte 2 % à 3 % de RM légers (QI entre 50 et 70) et 0,3 % à 0,4 % de RM sévères (QI < 50), avec une fréquence de 3 garçons pour 2 filles. Les RMLX ont quant à eux une incidence de l’ordre de 1,8/1 000 naissances masculines. Ils peuvent être classés en deux entités, qui aident à leur identification et orientent les recherches sur leurs mécanismes étiopathogéniques : ils peuvent ainsi être isolés, sans autres manifestations cliniques (RM non spécifiques, RMX), ou s’accompagner de signes permettant de les individualiser en syndromes parfois très caractéristiques (RM syndromiques, RMXS).
Chercher la femme
La transmission du phénotype RM, le plus souvent par des femmes vectrices, suit un mode récessif et seuls les garçons, hémizygotes pour le chromosome X, sont atteints sur plusieurs générations. L’analyse des familles révèle parfois une hérédité plus complexe, semi-dominante, avec des manifestations chez les femmes (cas de la maladie de l’X fragile). Enfin, des RM sporadiques observés chez des filles peuvent aussi relever d’une mutation dominante de novo, létale chez le garçon, d’un gène porté par l’X (exemple du syndrome de Rett).
De nouveaux gènes
Jamel Chelly a publié en 2000 une revue des RMLX ((→) m/s 2000, n° 3, p. 363) dans laquelle il rapportait l’identification de 26 gènes de RMXS et de 7 gènes de RMX. Des données plus récentes décrivent aujourd’hui 38 gènes responsables de RMXS, sur les 138 entités décrites, et 20 gènes impliqués dans les RMX [1]. Au-delà de ces chiffres illustrant les progrès issus des données du séquençage du génome humain et des efforts conjoints de plusieurs laboratoires européens pour identifier ces gènes (http://www.euromrx.com), les découvertes de ces cinq dernières années ont confirmé le grand nombre de gènes impliqués dans les RMLX et la diversité des fonctions biologiques exercées par les protéines leur correspondant. Les études soulignent également la grande hétérogénéité allélique et phénotypique de ces RMLX, illustrée notamment par des mutations du gène ARX responsables à la fois de RMXS et de RMX. Par ailleurs, des gènes initialement classés comme responsable de RMX (notamment le gène de l’oligophrénine 1) ont été reclassés en gènes responsables de RMXS, après un nouvel examen clinique des patients. Face à une telle complexité, la limite entre les formes syndromiques et non spécifiques est parfois remise en question, ce qui devrait rendre de plus en plus difficile le classement de ces gènes dans l’une ou l’autre des deux catégories de retards mentaux liés à l’X.
La description de l’ensemble de ces gènes et des pathologies qui résultent de leurs mutations dépasse de beaucoup le cadre de cette synthèse : nous limiterons donc celle-ci aux gènes les plus fréquents, ou les plus représentatifs de la complexité des RMLX.
Mutations du gène de la FMRP : syndrome de l’X fragile
Le syndrome de l’X fragile (OMIM #309550) est la cause la plus fréquente de RM héréditaire. Celui-ci est le plus souvent modéré [2, 3], et l’examen neuro-anatomique de cerveaux de patients a mis en évidence des défauts morphologiques au niveau des épines dendritiques (compartiments post-synaptiques de la plupart des neurones excitateurs) [4]. Ces observations suggèrent qu’un défaut de connexion entre les neurones puisse limiter la transmission synaptique, le RM étant alors l’expression clinique de ces limitations.
Le mécanisme pathogénique de l’X fragile a déjà été décrit dans ces colonnes ((→) m/s 1991, n° 4, p. 378 et 1996, n° 3, p. 415).
Les patients atteints d'X fragile ont une expansion du gène FMR1 qui dépasse 200 répétitions (mutation complète), conduisant à une hyperméthylation de la région promotrice et à l’extinction de l’expression de ce gène codant pour la protéine FRMP (fragile X mental retardation protein). Bien que les porteurs d’une prémutation ne soient pas atteints d’X fragile, ces allèles intermédiaires sont associés à d’autres phénotypes tels que la ménopause précoce ou une nouvelle forme de maladie neurodégénérative, le FXTAS (fragile X tremor ataxia syndrome) (voir Encadré) [5] ; chez ces patients, la quantité de protéines FMRP est diminuée, tandis que le taux d’ARNm correspondant est plus élevé : il semblerait donc que ces nouvelles pathologies soient liées à la surexpression des ARNm porteurs de la prémutation, leur conférant ainsi une fonction pathogénique (Figure 1).
Figure 1
Modèle hypothétique de l’effet toxique des ARNm prémutés de la protéine FMRP chez les patients atteints de FXTAS.
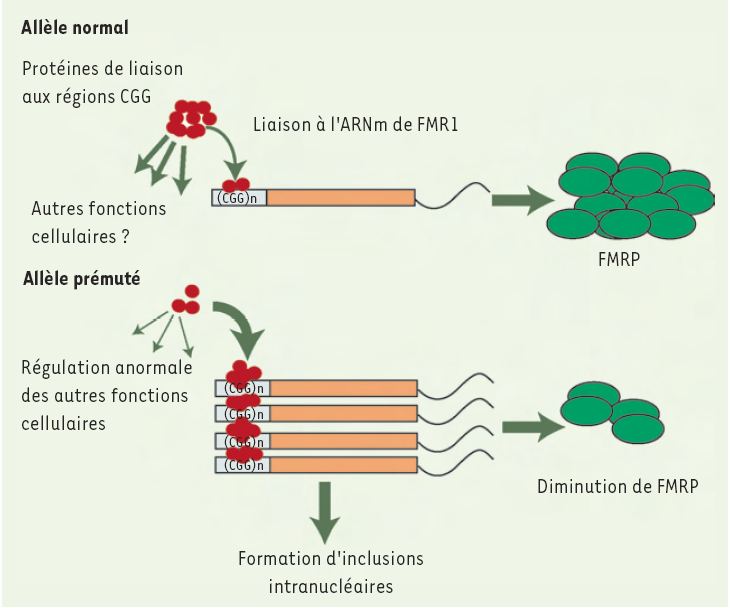
Dans le cas d’allèles normaux, des protéines de liaison des CGG se fixent sur la région 5’ non codante de l’ARNm de FMR1 et participent à la régulation de son expression ; ces facteurs seraient également capables de réguler l’expression d’autres transcrits. L’accumulation des ARNm prémutés chez les sujets atteints de FXTAS conduirait, quant à elle, à une séquestration de ces facteurs protéiques capables de lier les régions CGG : ces protéines seraient alors déplétées de la réserve cellulaire et ne seraient plus disponibles pour exercer leur fonction régulatrice normale sur d’autres ARNm. L’accumulation de ces ARNm prémutés conduirait également à la formation d’inclusions intranucléaires. FMRP : fragile X mental retardation protein ; FXTAS : fragile X tremor ataxia syndrome (d’après [5]).
L’analyse fonctionnelle de la protéine FMRP a montré qu’elle s’associe aux polyribosomes et peut se lier à certains ARNm dont elle régule la traduction, notamment au niveau des synapses [6, 7]. Le mécanisme de l’inhibition n’est pas encore connu, mais il a été montré que la protéine de drosophile dFMRP se lie au complexe RISC (RNA-induced silencing complex) impliqué dans les mécanismes d’interférence par l’ARN, conduisant soit à la dégradation de l’ARNm cible, soit à l’inhibition de sa traduction [8].
Par ailleurs, la recherche de protéines interagissant avec FMRP a permis l’identification de deux protéines cytoplasmiques, CYFIP (cytoplasmic FMRP interacting proteins) 1 et 2, elles-mêmes capables de lier la forme activée de la GTPase Rac1. Une voie de signalisation régulée par la GTPase Rac1, responsable du remodelage du cytosquelette et de la synthèse protéique via CYFIP et dFMRP, a d’ailleurs été mise évidence chez la drosophile [9, 10] (Figure 2). Or la synthèse locale de protéines dans les épines dendritiques, en réponse à l’activité de la synapse, est importante pour induire la stabilité de cette dernière, et la protéine FMRP pourrait contrôler localement la synthèse de protéines en modulant la traduction de certains messagers. Ainsi, l’absence de FMRP chez certains sujets conduirait à une dérégulation de la synthèse protéique : les synapses stimulées ne pourraient être stabilisées, et les épines dendritiques adopteraient alors la morphologie immature observée.
Figure 2
Voie de signalisation cellulaire relayée par les GTPases Rho (Ras homolog).
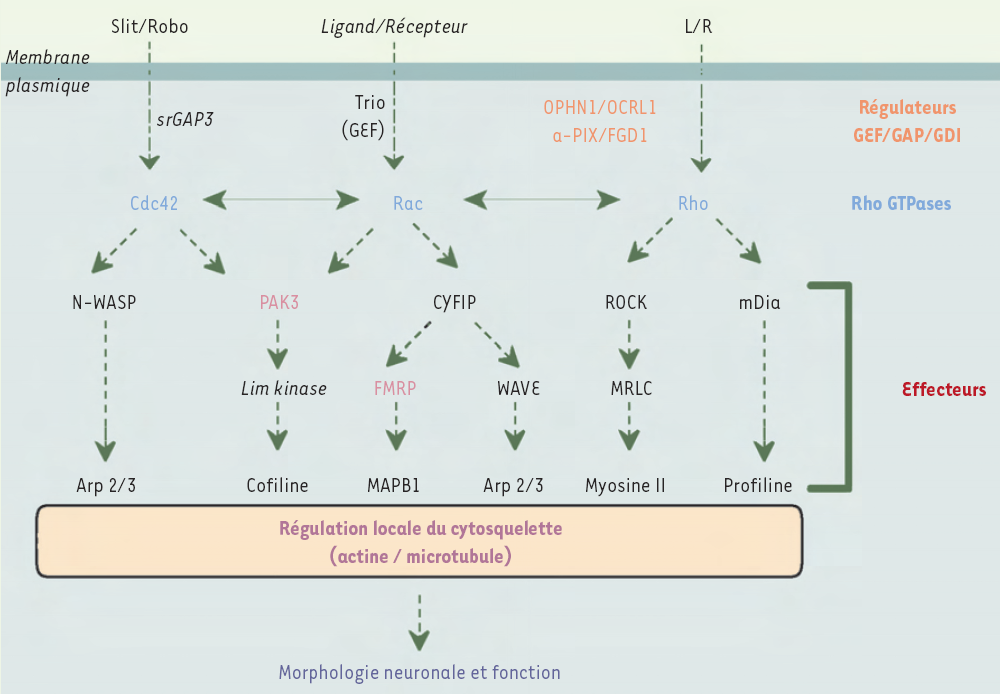
Les petites protéines G oscillent entre deux états, l’un inactif (liant le GDP) et l’autre actif (liant le GTP). Après stimulation du récepteur par son ligand, les échanges GDP/GTP sont régulés par des protéines GEF (guanine-nucleotide exchange factor), GAP (GTPases activating protein) et GDI (guanine-nucleotide-dissociation factor). Sous leur forme activée, les GTPases relaient le signal vers des protéines effectrices conduisant, en autres fonctions cellulaires, au remodelage des cytosquelettes d’actine et des microtubules. Le déficit en OPHN1, OCRL1, α-PIX, FGD1, PAK3 ou FMRP pourrait avoir des effets délétères sur la morphologie neuronale et la transmission synaptique. OPHN1 : oligophrénine 1 ; OCRL1 : de « oculocerebrorenal syndrome of Lowe » ; α-PIX : PAK-interacting exchange factor ; FGD1 : de « faciogenital dysplasia » ; PAK3 : p21-activated kinase 3 ; FMRP : fragile X mental retardation protein.
Mutations du gène de la protéine ARX : un exemple d’hétérogénéité poussée à l’extrême
La recherche de gènes impliqués dans des RMX, localisés autour de la région Xp22.1, a permis d’impliquer un nouveau gène, ARX (Aristaless related homeobox, du fait de son homologie avec la protéine de drosophile aristaless al) [11]. Des mutations de ce gène ont été découvertes chez des garçons atteints, appartenant à 9 familles « RMX » (pour 7 d’entre elles, la mutation avait été préalablement localisée en Xp22.1,2), et dans 1 cas sporadique. Certaines de ces mutations, qui sont des duplications/insertions en phase par rapport au cadre ouvert de lecture, conduisent à l’expansion de répétitions alanine au niveau de la protéine. Ces expansions de poly-alanine, qui rappellent celles de poly-glutamine dans certaines maladies neurodégénératives, pourraient conduire à la formation d’agrégats toxiques pour la cellule, responsables de la mort cellulaire ; les résultats des travaux testant cette hypothèse en surexprimant des protéines ARX mutées sont toutefois discordants, et la question reste ouverte [12, 13].
Outre l’implication du gène ARX dans les RMX, des mutations faux-sens ou des expansions de poly-alanine ont également été identifiées dans différentes formes syndromiques de RM (RMXS) : le syndrome de West (OMIM #308350), caractérisé par des convulsions s’accompagnant d’un tracé d’hypsarhythmie à l’électro-encéphalogramme et d’un RM, le syndrome de Partington (OMIM #309510), dans lequel on observe un RM et des mouvements dystoniques des mains, et des formes de RM associées à des épilepsies myocloniques et à une spasticité [14] ; les patients atteints de l’un ou l’autre de ces syndromes ne présentent pas de malformations cérébrales. Les mutations plus sévères du gène ARX, s’accompagnant notamment d’une perte totale de fonction de la protéine codée, sont quant à elles associées au syndrome XLAG (X-linked lissencephaly with ambiguous genitalia, OMIM#300215), caractérisé par une lissencéphalie liée à l’X et des malformations génitales [15].
À cette évidente hétérogénéité phénotypique multi-allélique des mutations du gène ARX s’ajoute une hétérogénéité phénotypique mono-allélique et intrafamiliale. En effet, la même duplication de 24 paires de bases, responsable d’une extension de 8 alanines, est retrouvée indifféremment dans les RMX et dans certains des syndromes décrits ci-dessus, à l’exception du syndrome XLAG et des épilepsies myocloniques. Par rapport à la situation de la majorité des gènes impliqués dans les RMLX, pour lesquels une mutation a été identifiée au sein de quelques familles seulement (1 à 3), cette duplication de 24 paires de bases dans le gène ARX est relativement fréquente, puisqu’elle représente plus de 6,6 % des RMLX [11-16].
La protéine ARX est un facteur de transcription de la famille des protéines à homéodomaines, dont les gènes cibles ne sont pas encore connus, mais qui est majoritairement exprimée au niveau du testicule et du cerveau. Le modèle murin correspondant à une perte totale de la protéine ARX est létal quelques jours après la naissance : les souriceaux présentent des anomalies sévères liées à un défaut de prolifération des neuroblastes, ainsi que des anomalies de migration/différenciation des interneurones GABAergiques [15]. Cette expression physiologique d’ARX dans les interneurones de type GABAergiques pourrait d’ailleurs expliquer la survenue de crises d’épilepsie chez les patients dont la protéine ARX est mutée.
Mutations du gène de l’oligophrénine 1 : de RMX à RMXS
Depuis son identification et son implication dans les RMX en 1998 [17], plusieurs mutations ont été identifiées dans le gène codant pour l’oligophrénine 1 (OPHN1), la majorité d’entre elles conduisant à une perte de la fonction de la protéine OPHN1. L’examen rétrospectif des patients a permis de définir des signes dysmorphiques et de caractériser par IRM une hypoplasie du cervelet, avec une agénésie des lobules VI et VII au niveau du vermis postérieur (Figure 3) [18-20]. En corollaire, une étude prospective récente, réalisée chez des patients atteints de RM avec hypoplasie cérébelleuse, montre que 12 % d’entre eux présentent une mutation dans le gène OPHN1 [21]. En quoi cette anomalie du cervelet contribue t-elle au RM ? Des hypoplasies du vermis postérieur ont également été retrouvées dans d’autres RM, dont le syndrome de Down et l’X fragile [22], ou encore chez des patients atteints d’autisme [23], suggérant que le cervelet pourrait être impliqué dans le fonctionnement cognitif.
Figure 3
Hypoplasie cérébelleuse chez les patients mutés dans le gène OPHN1.
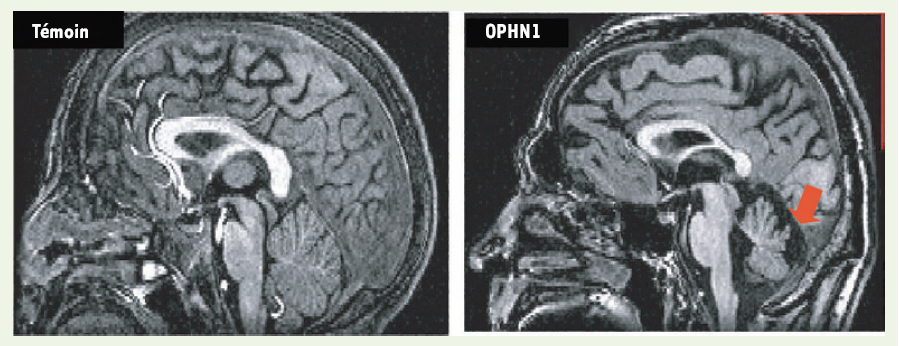
Reconstruction sagittale d’IRM 3D T1 d’un individu normal (à gauche) et d’un patient ayant une mutation dans le gène de l’oligophénine 1 (OPHN1) (à droite). À l’hypoplasie du cervelet s’ajoute une agénésie partielle du vermis postérieur au niveau des lobules VI et VII (flèche rouge).
Sur le plan physiopathologique, l’hypothèse initiale d’un défaut de morphologie neuronale [17] vient récemment d’être confortée par des études montrant un raccourcissement de la longueur des épines dendritiques après inactivation du gène OPHN1 par la stratégie de l’interférence par l’ARN [24]. Ainsi, un défaut de connexion neuronale serait à l’origine du RM, comme dans les cas d’X fragile ; cette similitude est d’autant plus remarquable que les protéines FMRP et OPHN1, tout comme quatre autres protéines codées par des gènes impliqués dans les RMX, semblent appartenir aux mêmes voies de signalisation relayées par les GTPases Rho, soit comme régulateurs (α-PIX, FGD1, OPHN1, OCRL1), soit comme effecteurs (PAK3, FMRP) (Figure 2). Les GTPases Rho, via la régulation des cytosquelettes d’actine ou des microtubules, contrôlent l’ensemble des étapes du développement neuronal, de la division jusqu’à la maturation des synapses ; elles participent également, au cours de la vie adulte, à la stabilité et au remodelage des synapses, mécanismes essentiels à la plasticité synaptique [25] ; enfin, elles sont directement impliquées dans le fonctionnement des synapses au cours des processus d’exocytose ou d’endocytose des vésicules synaptiques. Pour certaines de ces protéines (FMRP, OPHN1), les résultats fonctionnels obtenus orientent les hypothèses physiopathologiques de ces RMX vers un défaut de morphologie neuronale ; il est en revanche difficile d’extrapoler ce mécanisme à l’ensemble des autres protéines précitées, étant donné la diversité des fonctions biologiques relayées par les GTPases.
Outre l’anomalie du cervelet observée chez les patients déficients en protéine OPHN1, les données obtenues soulignent l’importance de la réalisation d’un nouvel examen clinique, biochimique et radiologique dans les familles RMX pour lesquelles une mutation a été trouvée dans le même gène (homogénéité moléculaire), afin d’identifier une ou plusieurs anomalies spécifiques communes permettant de définir un nouveau syndrome.
Conclusions
Ces dernières années ont vu l’identification d’un grand nombre de gènes (environ 60) de RMLX, toutes formes confondues. Toutefois, dans de nombreux cas familiaux dont le phénotype est clairement lié à l’X (58 sur les 80 familles RMX, par exemple), aucune mutation n’a été trouvée, soit parce que la recherche de mutations n’a pas été systématiquement effectuée pour tous les gènes RMLX connus dans un intervalle génétique donné, soit parce qu’elles affectent de nouveaux gènes non encore identifiés. Ainsi, le nombre total de gènes impliqués dans les RMLX est difficile à estimer à ce jour, et pourrait varier du simple au double (jusqu’à 100) [1-16]. À l’avenir, les efforts devraient porter non seulement sur la recherche systématique de mutations dans tous les gènes d’un intervalle donné, mais aussi sur la caractérisation des remaniements chromosomiques ou subchromosomiques, comme les translocations X ; autosomes ou les microdélétions : ces approches sont en effet toujours d’actualité, car elles permettent de restreindre les recherches à une petite région.
Bien que la classification conventionnelle des RM en « syndromiques (RMXS) » et « non spécifiques (RMX) » soit très utile, pour des raisons médicales et génétiques évidentes, l’extrême hétérogénéité des RMLX conduit à évoluer vers d’autres modes de classification plus simples, reposant notamment sur la présence ou non d’anomalies du cerveau détectables par imagerie ; ces anomalies, spécifiques ou non, sont le plus souvent sévères (lissencéphalie, agénésie partielle, hypoplasie, holoprocencéphalie…), et surviennent au cours du développement : le RM apparaît alors secondaire à la désorganisation du SNC. À l’inverse, les gènes impliqués dans les RM dénués d’anomalies cérébrales détectables participent directement à l’établissement ou au maintien des fonctions cognitives : l’étude des protéines correspondantes et de leurs fonctions devrait permettre une meilleure appréhension de la cognition, et nous devrions évoluer vers une nouvelle classification des RMLX fondée sur une approche plus moléculaire. Ainsi, en classant les gènes actuels en fonction de l’activité biologique de leur produit ou d’un mécanisme physiopathologique commun, on peut distinguer les protéines impliquées dans des voies de métabolismes, celles qui s’associent au cytosquelette d’actine ou des microtubules, celles qui contrôlent leur dynamique en participant à des voies de signalisation ou encore celles qui régulent l’expression génique [26]. Aucun mode de classification des RMLX ne permet donc à l’heure actuelle de les différencier de manière univoque, sans trouver d’exceptions ; chaque classification (clinique, génétique, fonctionnelle ou physiopathologique) présente des avantages et des inconvénients, en fonction de l’intérêt de chacun.
Appendices
Références
- 1. Ropers HH, Hamel BC. X-linked mental retardation. Nat Rev Genet 2005 ; 6 : 46-57.
- 2. Hagerman RJ, Silverman AC. Fragile X syndrome : diagnosis, treatment, and research. Baltimore : Johns Hopkins University Press, 1991 ; XIV : 378 p.
- 3. Fisch GS, Simensen R, Tarleton J, et al. Longitudinal study of cognitive abilities and adaptive behavior levels in fragile X males : a prospective multicenter analysis. Am J Med Genet 1996 ; 64 : 356-61.
- 4. Irwin SA, Galvez R, Greenough WT. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex 2000 ; 10 : 1038-44.
- 5. Hagerman PJ, Hagerman RJ. The fragile-X premutation : a maturing perspective. Am J Hum Genet 2004 ; 74 : 805-16.
- 6. Bardoni B, Mandel JL. Advances in understanding of fragile X pathogenesis and FMRP function, and in identification of X linked mental retardation genes. Curr Opin Genet Dev 2002 ; 12 : 284-93.
- 7. Zalfa F, Giorgi M, Primerano B, et al. The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell 2003 ; 112 : 317-27.
- 8. Ishizuka A, Siomi MC, Siomi H. A Drosophila fragile X protein interacts with components of RNAi and ribosomal proteins. Genes Dev 2002 ; 16 : 2497-508.
- 9. Schenck A, Bardoni B, Langmann C, et al. CYFIP/Sra-1 controls neuronal connectivity in Drosophila and links the Rac1 GTPase pathway to the fragile X protein. Neuron 2003 ; 38 : 887-98.
- 10. Billuart P, Chelly J. From fragile X mental retardation protein to Rac1 GTPase : new insights from Fly CYFIP. Neuron 2003 ; 38 : 843-5.
- 11. Bienvenu T, Poirier K, Friocourt G, et al. ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Hum Mol Genet 2002 ; 11 : 981-91.
- 12. Poirier K, Van Esch H, Friocourt G, et al. Neuroanatomical distribution of ARX in brain and its localisation in GABAergic neurons. Brain Res Mol Brain Res 2004 ; 122 : 35-46.
- 13. Nasrallah IM, Minarcik JC, Golden JA. A polyalanine tract expansion in Arx forms intranuclear inclusions and results in increased cell death. J Cell Biol 2004 ; 167 : 411-6.
- 14. Stromme P, Mangelsdorf ME, Scheffer IE, Gecz J. Infantile spasms, dystonia, and other X-linked phenotypes caused by mutations in Aristaless related homeobox gene, ARX. Brain Dev 2002 ; 24 : 266-8.
- 15. Kitamura K, Yanazawa M, Sugiyama N, et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet 2002 ; 32 : 359-69.
- 16. Mandel JL, Chelly J. Monogenic X-linked mental retardation : is it as frequent as currently estimated ? The paradox of the ARX (Aristaless X) mutations. Eur J Hum Genet 2004 ; 12 : 689-93.
- 17. Billuart P, Bienvenu T, Ronce N, et al. Oligophrenin-1 encodes a rhoGAP protein involved in X-linked mental retardation. Nature 1998 ; 392 : 923-6.
- 18. Bergmann C, Zerres K, Senderek J, et al. Oligophrenin 1 (OPHN1) gene mutation causes syndromic X-linked mental retardation with epilepsy, rostral ventricular enlargement and cerebellar hypoplasia. Brain 2003 ; 126 : 1537-44.
- 19. Philip N, Chabrol B, Lossi AM, et al. Mutations in the oligophrenin-1 gene (OPHN1) cause X linked congenital cerebellar hypoplasia. J Med Genet 2003 ; 40 : 441-6.
- 20. Des Portes V, Boddaert N, Sacco S, et al. Specific clinical and brain MRI features in mentally retarded patients with mutations in the Oligophrenin-1 gene. Am J Med Genet 2004 ; 124A : 364-71.
- 21. Zanni G, Saillour Y, Nagara M, et al. OPHN1 mutations are a frequent cause of X-linked mental retardation with cerebellar hypoplasia. Neurology 2005 (sous presse).
- 22. Mostofsky SH, Mazzocco MM, Aakalu G, et al. Decreased cerebellar posterior vermis size in fragile X syndrome : correlation with neurocognitive performance. Neurology 1998 ; 50 : 121-30.
- 23. Kaufmann WE, Cooper KL, Mostofsky SH, et al. Specificity of cerebellar vermian abnormalities in autism : a quantitative magnetic resonance imaging study. J Child Neurol 2003 ; 18 : 463-70.
- 24. Govek EE, Newey SE, Akerman CJ, et al. The X-linked mental retardation protein oligophrenin-1 is required for dendritic spine morphogenesis. Nat Neurosci 2004 ; 7 : 364-72.
- 25. Luo L. Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci 2000 ; 1 : 173-80.
- 26. Renieri A, Pescucci C, Longo I, et al. Non-syndromic X-linked mental retardation : from a molecular to a clinical point of view. J Cell Physiol 2005 ; 240 : 8-20.
List of figures
Figure 1
Modèle hypothétique de l’effet toxique des ARNm prémutés de la protéine FMRP chez les patients atteints de FXTAS.
Dans le cas d’allèles normaux, des protéines de liaison des CGG se fixent sur la région 5’ non codante de l’ARNm de FMR1 et participent à la régulation de son expression ; ces facteurs seraient également capables de réguler l’expression d’autres transcrits. L’accumulation des ARNm prémutés chez les sujets atteints de FXTAS conduirait, quant à elle, à une séquestration de ces facteurs protéiques capables de lier les régions CGG : ces protéines seraient alors déplétées de la réserve cellulaire et ne seraient plus disponibles pour exercer leur fonction régulatrice normale sur d’autres ARNm. L’accumulation de ces ARNm prémutés conduirait également à la formation d’inclusions intranucléaires. FMRP : fragile X mental retardation protein ; FXTAS : fragile X tremor ataxia syndrome (d’après [5]).
Figure 2
Voie de signalisation cellulaire relayée par les GTPases Rho (Ras homolog).
Les petites protéines G oscillent entre deux états, l’un inactif (liant le GDP) et l’autre actif (liant le GTP). Après stimulation du récepteur par son ligand, les échanges GDP/GTP sont régulés par des protéines GEF (guanine-nucleotide exchange factor), GAP (GTPases activating protein) et GDI (guanine-nucleotide-dissociation factor). Sous leur forme activée, les GTPases relaient le signal vers des protéines effectrices conduisant, en autres fonctions cellulaires, au remodelage des cytosquelettes d’actine et des microtubules. Le déficit en OPHN1, OCRL1, α-PIX, FGD1, PAK3 ou FMRP pourrait avoir des effets délétères sur la morphologie neuronale et la transmission synaptique. OPHN1 : oligophrénine 1 ; OCRL1 : de « oculocerebrorenal syndrome of Lowe » ; α-PIX : PAK-interacting exchange factor ; FGD1 : de « faciogenital dysplasia » ; PAK3 : p21-activated kinase 3 ; FMRP : fragile X mental retardation protein.
Figure 3
Hypoplasie cérébelleuse chez les patients mutés dans le gène OPHN1.
Reconstruction sagittale d’IRM 3D T1 d’un individu normal (à gauche) et d’un patient ayant une mutation dans le gène de l’oligophénine 1 (OPHN1) (à droite). À l’hypoplasie du cervelet s’ajoute une agénésie partielle du vermis postérieur au niveau des lobules VI et VII (flèche rouge).