Article body
La division des cellules eucaryotes est réglée par des complexes cycline-CDK (kinases dépendantes des cyclines) qui contrôlent la progression des cellules dans chaque phase du cycle cellulaire (Figure 1). Ces CDK sont, comme leur nom l’indique, en partie activées par leur association aux sous-unités régulatrices, les cyclines, dont l’abondance varie au cours du cycle cellulaire. L’activité de ces complexes est inhibée par deux familles d’inhibiteurs (CKI) : les CKI de la famille des Ink4 inhibent CDK4 et CDK6 ; ceux de la famille Cip/Kip inhibent l’activité de tous les complexes cycline-CDK [1]. La progression des cellules de mammifères en phase G1, dépendante des signaux mitogènes, est contrôlée par l’association des cyclines D (D1, D2 et D3) aux kinases CDK4 et CDK6, ces complexes phosphorylant la protéine du rétinoblastome pRB (Figure 1) [1].
Figure 1
Cycle cellulaire chez les mammifères et complexes cyclines-CDK.
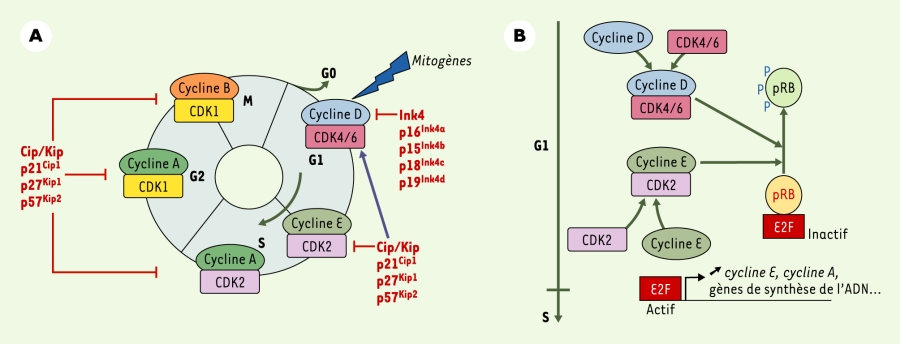
A. Le cycle cellulaire est composé de quatre phases G1, S, G2 et M. La progression entre chaque phase est contrôlée par différents complexes cycline-CDK. L’induction de l’expression des cyclines D par des signaux mitogènes est nécessaire à la progression des cellules en début de phase G1. L’activité des complexes cycline-CDK est inhibée par deux familles d’inhibiteurs de CDK : la famille des protéines Ink4 (p16Ink4a, p15Ink4b, p18Ink4c et p19Ink4d) est spécifique des complexes cycline D-CDK4/6 ; les protéines de la famille Cip/Kip (p21Cip1, p27Kip1 et p57Kip2) agissent sur tous les complexes cycline-CDK. L’association de p27Kip1 ou p21Cip1 aux complexes cycline D-CDK4/6 libère les complexes cycline E-CDK2, alors actifs. B. Dans les cellules de mammifères, la progression des cellules en début de phase G1, dépendante des signaux mitogènes, est contrôlée par l’association des cyclines D (D1, D2 et D3) aux kinases CDK4 et CDK6. Quand ils sont actifs, les complexes cycline D-CDK4/6 déclenchent la phosphorylation de la protéine du rétinoblastome (pRB), poursuivie en fin de phase G1 par les complexes cycline E-CDK2. Sous sa forme hyperphosphorylée, pRB est inactive et libère le facteur de transcription E2F, permettant ainsi l’expression des gènes essentiels à la transition G1-S dont le gène cycline E.
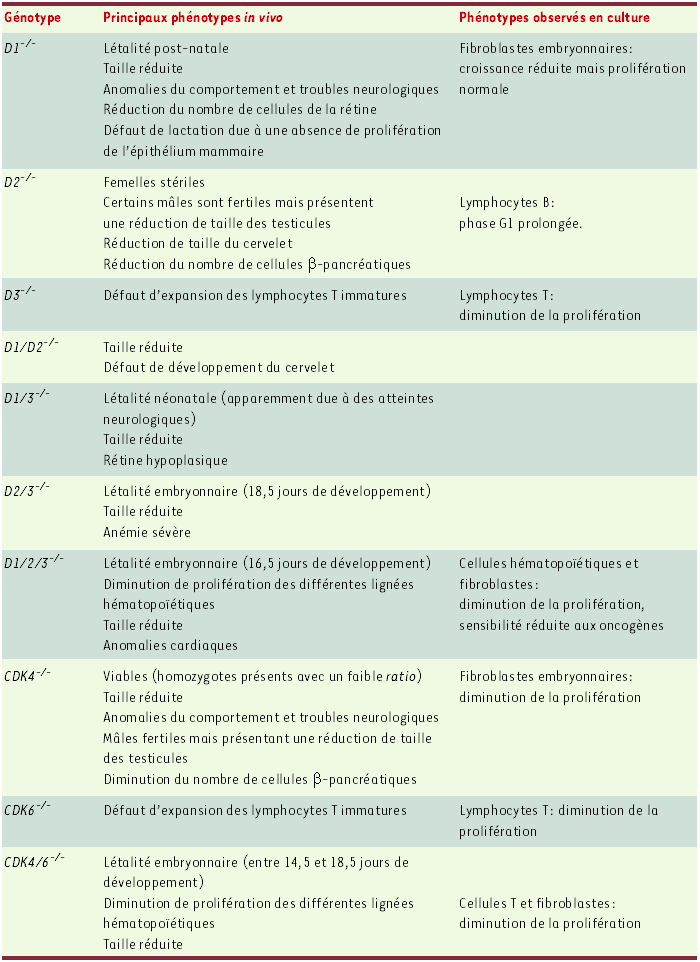
Leur expression étant sensible aux signaux mitogènes [1], il est admis que les cyclines D couplent la signalisation extracellulaire à la machinerie du cycle. Afin d’étudier in vivo la fonction des cyclines D et de CDK4/6, des lignées murines invalidées pour chacun de ces gènes avaient été établies [2-5]. Ces souris sont pour la plupart viables, les phénotypes observés étant restreints à certains organes (Tableau I). Ces données suggéraient une redondance fonctionnelle entre les cyclines D1, D2 et D3 et entre CDK4 et CDK6 mais elles n’avaient pas permis de préciser leur rôle physiologique, éventuellement spécifique d’un tissu, ni de savoir si ces cyclines et CDK étaient les seuls médiateurs entre la signalisation extracellulaire et le cycle cellulaire.
Tableau I
Phénotypes des mutants dont les gènes codant pour les cyclines D1, D2 et D3 et CDK4 et CDK6 ont été invalidés.
(d’après [10])
Récemment, l’obtention de souris invalidées pour les trois cyclines D (D1/2/3-/-) ou pour CDK4 et CDK6 (CDK4/6-/-) a apporté de nouveaux éléments de réponse à ces questions [4, 6]. Les premières meurent à 16,5 jours et les secondes à 18,5 jours de développement. Dans les deux cas, les embryons ont une taille réduite mais la majorité des organes ont une morphogenèse normale. Seul un défaut cardiaque est décrit pour les embryons D1/2/3-/-. Cette létalité embryonnaire est due à une anémie sévère consécutive à un défaut majeur d’hématopoïèse, lié à une diminution du taux de prolifération spécifique des cellules hématopoïétiques (Tableau I). Ainsi, il est surprenant de constater que, dans ces embryons, seules les cellules du lignage hématopoïétique prolifèrent de façon dépendante des complexes cycline D-CDK4/6, la majorité des tissus étant indépendants de leur activité.
Les complexes cycline D-CDK4/6 étant considérés comme les capteurs de la signalisation extracellulaire lors de la ré-entrée dans le cycle des cellules quiescentes, la réponse aux signaux extracellulaires de fibroblastes dérivés des deux types d’embryons mutants a été étudiée. En culture, ces cellules prolifèrent, mais moins activement que des cellules sauvages. Elles répondent cependant à une stimulation mitogénique, puisque, arrêtées en phase de quiescence (G0) par privation en sérum, elles ont une cinétique d’entrée en phase S, après stimulation, similaire à celle de cellules sauvages.
D’un point de vue moléculaire, M. Malumbres et al. montrent que dans les cellules CDK4/6-/-, un complexe cycline D-CDK2 se forme et est, en partie, responsable de la phosphorylation de pRB, permettant ainsi la prolifération de ces cellules [4]. Cependant, les auteurs précisent que leurs expériences ne sont pas suffisantes pour prouver une compensation totale de la perte des activités CDK4 et CDK6 par CDK2. K. Kozar et al. montrent que les fibroblastes cycline D1/2/3-/- ont une prolifération dépendante de CDK2 [6]. Dans ces cellules, après stimulation sérique, les complexes cycline E- et cycline A-CDK2 sont activés avec une cinétique identique à celle observée dans les cellules sauvages et l’activité de ces complexes suffit à inactiver pRB. Dans les deux types d’invalidations étudiées, les cellules entrent en phase S malgré une phosphorylation incomplète de pRB.
Ainsi, ces études montrent que les complexes cycline D-CDK4/6 ne sont pas indispensables à la prolifération et à l’entrée dans le cycle cellulaire de cellules quiescentes et révèlent l’existence d’un mécanisme indépendant des cyclines D, capable de coupler l’activation des complexes impliquant la kinase CDK2 à la signalisation extracellulaire. Néanmoins, l’absence des cyclines D se traduit tout de même par une susceptibilité réduite des cellules à une transformation oncogénique par Ras et Myc. De plus, le modèle actuellement admis pour expliquer la progression en phase G1 suggère une séquestration de l’inhibiteur p27Kip1 par les complexes cycline D-CDK4/6 (Figure 1). Or, dans les cellules mutantes D1/2/3-/- et CDK4/6-/-, les concentrations de p21Cip1 et p27Kip1 sont diminuées et l’association de p27Kip1 à CDK2 n’est apparemment pas modifiée.
Ces résultats bouleversent donc les modèles établis, puisque, de façon inattendue, les mutants D1/2/3-/- et CDK4/6-/- ne montrent pas d’anomalie majeure de la prolifération. Au contraire, l’étude de ces mutants montre que la majorité des cellules de mammifères peut proliférer indépendamment des complexes cycline D-CDK4/6. La nature des mécanismes compensateurs et leur implication dans l’intégration des signaux extracellulaires avec le contrôle de la prolifération restent maintenant à élucider.
Ces études révèlent également l’existence d’une spécificité de fonction de CDK4/6 et des cyclines D dans les cellules hématopoïétiques. Or, chez la souris et le poulet, on sait que les cyclines D présentent une expression régionalisée et dynamique au cours du développement [7-9]. Les analyses phénotypiques des souris invalidées pour CDK4/6 et les cyclines D ayant été réalisées à des stades tardifs du développement, des phénomènes compensatoires ont pu se produire. L’étude de stades plus précoces du développement ainsi que l’inactivation ciblée dans le temps de ces gènes devraient permettre de préciser les fonctions et les spécificités tissulaires de ces acteurs du cycle cellulaire.
Appendices
Remerciements
Je remercie le Pr B. Ducommun pour son aide à la rédaction du manuscrit.
Références
- 1. Sherr CJ, Roberts JM. CDK inhibitors : positive and negative regulators of G1-phase progression. Genes Dev 1999 ; 13 : 1501-12.
- 2. Rane SG, Dubus P, Mettus RV, et al. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in β-islet cell hyperplasia. Nat Genet 1999 ; 22 : 44-52.
- 3. Tsutsui T, Hesabi B, Moons DS, et al. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol Cell Biol 1999 ; 19 : 7011-9.
- 4. Malumbres M, Sotillo R, Santamaria D, et al. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 2004 ; 118 : 493-504.
- 5. Ciemerych MA, Kenney AM, Sicinska E, et al. Development of mice expressing a single D-type cyclin. Genes Dev 2002 ; 16 : 3277-89.
- 6. Kozar K, Ciemerych MA, Rebel VI, et al. Mouse development and cell proliferation in the absence of D-cyclins. Cell 2004 ; 118 : 477-91.
- 7. Lobjois V, Benazeraf B, Bertrand N, et al. Specific regulation of cyclins D1 and D2 by FGF and Shh signaling coordinates cell cycle progression, patterning, and differentiation during early steps of spinal cord development. Dev Biol 2004 ; 273 : 195-209.
- 8. Wianny F, Real FX, Mummery CL, et al. G1-phase regulators, cyclin D1, cyclin D2, and cyclin D3 : up-regulation at gastrulation and dynamic expression during neurulation. Dev Dyn 1998 ; 212 : 49-62.
- 9. Aguzzi A, Kiess M, Ruedi D, Hamel PA. Cyclins D1, D2 and D3 are expressed in distinct tissues during mouse embryogenesis. Transgenics 1996 ; 2 : 29-39.
- 10. Pagano M, Jackson PK. Wagging the dogma : tissue-specific cell cycle control in the mouse embryo. Cell 2004 ; 118 : 535-8.
List of figures
Figure 1
Cycle cellulaire chez les mammifères et complexes cyclines-CDK.
A. Le cycle cellulaire est composé de quatre phases G1, S, G2 et M. La progression entre chaque phase est contrôlée par différents complexes cycline-CDK. L’induction de l’expression des cyclines D par des signaux mitogènes est nécessaire à la progression des cellules en début de phase G1. L’activité des complexes cycline-CDK est inhibée par deux familles d’inhibiteurs de CDK : la famille des protéines Ink4 (p16Ink4a, p15Ink4b, p18Ink4c et p19Ink4d) est spécifique des complexes cycline D-CDK4/6 ; les protéines de la famille Cip/Kip (p21Cip1, p27Kip1 et p57Kip2) agissent sur tous les complexes cycline-CDK. L’association de p27Kip1 ou p21Cip1 aux complexes cycline D-CDK4/6 libère les complexes cycline E-CDK2, alors actifs. B. Dans les cellules de mammifères, la progression des cellules en début de phase G1, dépendante des signaux mitogènes, est contrôlée par l’association des cyclines D (D1, D2 et D3) aux kinases CDK4 et CDK6. Quand ils sont actifs, les complexes cycline D-CDK4/6 déclenchent la phosphorylation de la protéine du rétinoblastome (pRB), poursuivie en fin de phase G1 par les complexes cycline E-CDK2. Sous sa forme hyperphosphorylée, pRB est inactive et libère le facteur de transcription E2F, permettant ainsi l’expression des gènes essentiels à la transition G1-S dont le gène cycline E.
List of tables
Tableau I
Phénotypes des mutants dont les gènes codant pour les cyclines D1, D2 et D3 et CDK4 et CDK6 ont été invalidés.
(d’après [10])