Abstracts
Résumé
Le syndrome de Kallmann De Morsier est une maladie du développement embryonnaire qui associe un hypogonadisme central et une anosmie. Alors que des mutations du gène KAL1 codant pour l’anosmine-1, une protéine présente dans certaines matrices extracellulaires pendant l’organogenèse, avaient été mises en évidence dans la forme liée au chromosome X, des mutations du gène qui code pour FGFR1, l’un des récepteurs des fibroblast growth factors (FGF), ont été récemment identifiées dans une forme autosomique dominante de la maladie. Il reste à découvrir les autres gènes impliqués dans ce syndrome cliniquement et génétiquement hétérogène. Cependant, nous proposons dès à présent une hypothèse physiopathologique unificatrice pour rendre compte de l’aplasie des bulbes olfactifs qui caractérise ce syndrome.
Summary
Kallmann syndrome (KAL) associates hypogonadotropic hypogonadism and anosmia, i.e. a deficiency of the sense of smell. Anosmia is related to the absence or the hypoplasia of the olfactory bulbs. Hypogonadism is due to GnRH deficiency, and is likely to result from the failed embryonic migration of GnRH-synthesizing neurons. These cells normally migrate from the olfactory epithelium to the forebrain along the olfactory nerve pathway. Kallmann syndrome is genetically heterogeneous. The gene responsible for the X-chromosome linked form of the disease, KAL-1, has been identified in 1991. KAL1 encodes a ~95 kDa glycoprotein of unknown function, which is present locally in various extracellular matrices during the period of organogenesis. The recent finding that FGFR1 mutations are involved in an autosomal dominant form of Kallmann syndrome (KAL-2), combined to the analysis of mutant mouse embryos that no longer express Fgfr1 in the telencephalon, suggests that the disease results from a deficiency in FGF-signaling at the earliest stage of olfactory bulb morphogenesis. We propose that the role of the KAL1 gene product, the extracellular matrix protein anosmin-1, is to enhance FGF-signaling, and suggest that the gender difference in anosmin-1 dosage (because KAL1 partially escapes X-inactivation) explains the higher prevalence of the disease in males.
Article body
Douze ans après l’isolement du gène KAL1 responsable de la forme liée au chromosome X de la maladie, une nouvelle étape vient d’être franchie dans la compréhension de la pathogénie du syndrome de Kallmann De Morsier avec l’identification de mutations du gène codant pour FGFR1 (fibroblast growth factor receptor 1) (Figure 1) chez certains individus atteints d’une forme autosomique dominante de la maladie [1]. Ce syndrome associe l’absence de puberté spontanée, par défaut de production de la gonadolibérine (GnRH, hormone hypothalamique qui contrôle l’axe endocrinien de la reproduction) [2], à une absence plus ou moins complète d’odorat (anosmie), en rapport avec une anomalie de la formation des bulbes olfactifs mise en évidence par l’étude anatomoclinique de De Morsier [3]. Entre ces deux symptômes si différents, il existe en fait un lien qui tient au rapport topographique transitoire, au cours de la vie embryonnaire, entre les neurones qui synthétisent la GnRH et le système olfactif périphérique [4].
Figure 1
Activation de FGFR1 par ses ligands FGF en présence de protéoglycanes à héparane sulfate.

FGFR1 est un récepteur membranaire actif sous forme de dimère, les monomères étant inactifs. La région extracellulaire, qui fixe le ligand FGF, comporte trois domaines de type immunoglobuline (IgI, IgII et IgIII). Entre les deux premiers s’intercale un module très riche en résidus acides (région acide), dont le rôle serait d’empêcher l’activation spontanée du récepteur en l’absence de son ligand [33]. La région intracellulaire comprend deux domaines à activité tyrosine kinase (TK). La formation d’un complexe moléculaire tripartite entre FGF, FGFR et la chaîne héparane sulfate d’un protéoglycanne est nécessaire à la dimérisation du récepteur, qui est suivie de son activation par autophosphorylation (P) de résidus tyrosine (Y) dans la région intracellulaire de la molécule. Les phosphotyrosines stimulent en retour l’activité tyrosine kinase du récepteur ou servent de points d’ancrage à des molécules de la cascade de signalisation.
Une maladie génétiquement hétérogène, plus fréquente chez les garçons
Le caractère héréditaire de cette maladie est connu depuis la description par Kallmann de trois familles comportant plusieurs garçons atteints [5]. D’autres cas familiaux ont été rapportés par la suite, avec trois modes de transmission possibles : récessif lié au chromosome X, autosomique dominant ou, plus rarement, récessif. Cependant, les cas sporadiques sont de loin les plus fréquents. De plus, l’incidence de la maladie serait trois à cinq fois plus élevée chez les garçons (1:8000) que chez les filles. Le degré variable de l’hypogonadisme et de l’anosmie, même au sein d’une même famille, ainsi que la pénétrance incomplète de la maladie dans des familles où la transmission s’effectue selon le mode dominant, ont été soulignés.
Le gène KAL1, responsable de la forme liée au chromosome X de la maladie, a été localisé dans la région Xp22.3 puis identifié en 1991 [6-8], et diverses mutations ponctuelles ou délétions intragéniques ont été mises en évidence dans des cas familiaux [9, 10]. En revanche, les mutations de KAL1 sont rares dans les cas sporadiques [11]. Ainsi, il semble que la franche prédominance masculine de cette maladie ne puisse pas être expliquée par la prévalence de la forme liée au chromosome X. KAL1 code pour une glycoprotéine modulaire d’environ 95 kDa, l’anosmine-1 (Figure 2), qui est présente localement dans certaines matrices extracellulaires (membranes basales et matrices interstitielles) pendant l’organogenèse [12].
Figure 2
Structure de l’anosmine-1.

L’anosmine-1 est une protéine de la matrice extracellulaire. Elle est présente localement, dans divers tissus, pendant la période de l’organogenèse. Son absence est à l’origine de la forme liée au chromosome X du syndrome de Kallmann De Morsier. Elle comporte une région aminoterminale riche en résidus cystéine, au sein de laquelle on identifie un domaine d’environ 50 acides aminés, d’abord mis en évidence dans la protéine acide du petit lait ou whey acidic protein (WAP), et conservé dans toutes les protéines orthologues de l’anosmine-1 humaine connues à ce jour, y compris chez les invertébrés. Ce motif est également présent dans plusieurs petites protéines sécrétées qui ont une activité inhibitrice de protéases à sérine, mais une telle activité demeure hypothétique pour l’anosmine-1 (comme d’ailleurs pour la protéine WAP). Le reste de la protéine comprend quatre domaines semblables aux motifs répétés de type III de la fibronectine, suivis par un court fragment carboxyterminal riche en acides aminés basiques et en histidine. L’anosmine-1 se lie aux glycosaminoglycannes du type héparane sulfate, ce qui suggère qu’elle participe à la signalisation par les facteurs FGF.
Par une approche de clonage positionnel fondée sur l’analyse de délétions interstitielles du chromosome 8 (région 8p11-p12), nous avons montré que des mutations du gène FGFR1 sont responsables d’une forme dominante de la maladie [1, 10].
Diverses autres anomalies phénotypiques accompagnent parfois l’hypogonadisme et l’anosmie qui définissent le syndrome de Kallmann De Morsier [4]. Si certaines résultent de la perte simultanée de gènes voisins consécutive à une délétion chromosomique chez de rares individus [1, 8], la plupart sont directement liées à la mutation d’un des gènes responsables de cette maladie [1, 9]. Dans ce cas, ces anomalies peuvent être présentes dans différentes formes génétiques de la maladie, avec toutefois une prévalence variable. La diversité des anomalies phénotypiques qui sont communes aux formes KAL-1 et KAL-2 (Tableau I) plaide en faveur d’un rôle de l’anosmine-1 dans la signalisation cellulaire via FGFR1. Le mécanisme précis de la coopération entre ces protéines n’est pas connu. Cependant, une telle interaction pourrait expliquer la fréquence plus élevée de la maladie chez les garçons, si l’on fait l’hypothèse que la concentration locale de l’anosmine-1 joue, dans certains tissus, un rôle critique pour la signalisation par les FGF. En effet, le gène KAL1, bien que situé en dehors de la région pseudo-autosomique[1] du chromosome X, échappe partiellement au phénomène d’inactivation qui concerne l’un des deux chromosomes X dans chaque cellule somatique chez la femme [6, 13]. Il en résulte une inégalité de production de l’anosmine-1 entre les deux sexes, en faveur du sexe féminin. Ainsi, une concentration locale d’anosmine-1 physiologiquement plus élevée chez les femmes pourrait-elle compenser, chez certaines, une situation pathologique d’haplo-insuffisance pour le gène FGFR1. Deux observations viennent à l’appui de cette hypothèse. Dans quatre des cinq familles atteintes de la forme KAL-2 chez lesquelles nous avons pu étudier la transmission de la mutation de FGFR1 à la dernière génération, c’est la mère, asymptomatique, qui était porteuse de la mutation [1]. Par ailleurs, alors que les souris génétiquement déficientes en Fgfr1 dans le télencéphale ont, comme dans la maladie humaine, une aplasie des bulbes olfactifs (voir plus loin) et meurent peu après la naissance, incapables de s’alimenter du fait de l’anosmie, ce phénotype n’est observé que chez les souris mutantes homozygotes. Les souris, mâles et femelles, hétérozygotes pour le gène inactivé sont viables et fertiles [14]. Or, à la différence du gène KAL1 des primates, le gène Kal1 murin est vraisemblablement situé dans la région pseudo-autosomique des chromosomes sexuels, et deux allèles sont donc exprimés dans l’un et l’autre sexe ; cela pourrait expliquer la différence de phénotype entre l’homme et la souris lorsqu’il n’existe qu’un seul allèle fonctionnel de FGFR1 ou Fgfr1. Si cette hypothèse est exacte, on devrait retrouver le phénotype anormal chez des souris double-hétérozygotes Fgfr1+/-, Kal1+/-.
Tableau I
Formes génétiques identifiées du syndrome de Kallmann De Morsier.

FGF : fibroblast growth factors ; FGFR1 : fibroblast growth factor receptor 1.
La pathogénie de l’anosmie du syndrome de Kallmann De Morsier : une hypothèse unificatrice…
La découverte, dans différentes espèces animales, que les neurones synthétisant la GnRH migrent pendant la vie embryonnaire en suivant le trajet des nerfs olfactifs [15] a apporté un éclairage nouveau à la physiopathologie de la maladie. Dans l’espèce humaine, ces neurones quittent la partie médiane de l’épithélium olfactif dès la 6e semaine de vie foetale [16]. Parvenus à la base du télencéphale, ils pénètrent dans le cerveau juste en arrière des futurs bulbes olfactifs, puis cheminent à la face médiane des hémisphères cérébraux jusqu’à la région hypothalamique, où se produira la neurosécrétion. Dans leur trajet extracérébral, et d’ailleurs sans doute aussi intracérébral, ces cellules migrent en étroite association avec les fibres du nervus terminalis (nerf crânien zéro), qui relie l’épithélium olfactif à la région hypothalamique et dont le rôle physiologique demeure inconnu [17].
En 1989, l’étude histopathologique d’un foetus masculin de 19 semaines porteur d’une délétion chromosomique incluant le gène KAL1 a montré, outre l’absence des bulbes olfactifs, une accumulation des neurones à GnRH dans la région nasale haute, au sein de terminaisons nerveuses abortives correspondant aux fibres du nerf olfactif et du nervus terminalis, tandis qu’aucun neurone à GnRH n’était détectable dans le cerveau [18]. Cette observation, certes postérieure à « l’accident » du développement ayant entraîné l’absence des bulbes olfactifs, suggérait néanmoins la possibilité d’un défaut primaire de l’élongation terminale des axones des neurones olfactifs, au moment où ces axones pénètrent dans le cerveau. Dans cette hypothèse, l’absence de développement des bulbes olfactifs serait secondaire à l’absence d’un contact stable entre les terminaisons des axones olfactifs et le cerveau, contact dont on sait depuis longtemps qu’il est nécessaire à la morphogenèse des bulbes olfactifs [19].
L’étude du profil d’expression précoce de l’anosmine-1 dans le système olfactif a révélé que durant la 6e semaine du développement embryonnaire, la protéine est présente dans la matrice interstitielle des bulbes olfactifs présomptifs, au pôle rostral du télencéphale [12]. A ce stade, la protéine n’est détectée ni dans l’épithélium sensoriel olfactif, ni sur le trajet extracérébral des fibres du nerf olfactif (Figure 3). Un tel profil d’expression était certes compatible avec un rôle de l’anosmine-1 sur le court trajet intracérébral des axones des neurones olfactifs primaires, mais il ne permettait pas non plus d’exclure une implication directe de la protéine dans les étapes initiales de la morphogenèse du bulbe olfactif, vers la fin de la 6e semaine. C’est cette dernière éventualité qui se trouve actuellement privilégiée, depuis la mise en évidence des mutations de FGFR1 dans une forme dominante du syndrome de Kallmann De Morsier et l’hypothèse d’une participation de l’anosmine-1 à la signalisation par les FGF [1, 10]. En effet, l’étude de souris génétiquement déficientes en Fgfr1 a montré que le contact initial des axones olfactifs avec la région rostrale du télencéphale avait lieu normalement. En revanche, l’évagination des bulbes olfactifs, qui survient normalement quelques heures après l’établissement de cette connexion nerveuse, ne se produit pas chez les souris mutantes homozygotes. Il semble que l’absence de Fgfr1 chez ces souris freine la différenciation en neuroblastes des cellules neuroépithéliales du télencéphale situées au voisinage des terminaisons axonales, c’est-à-dire la sortie de ces cellules du cycle mitotique [14], dont on pense qu’elle entraîne l’évagination des bulbes olfactifs du fait d’une réduction locale de la prolifération cellulaire [19] (Figure 4). Dès que l’orthologue de KAL1 aura été identifié chez la souris, l’inactivation du gène dans cette espèce devrait permettre d’étayer l’hypothèse unificatrice que nous proposons en montrant, chez les souris homozygotes mutantes, qu’il existe également un contact primaire des fibres olfactives avec le télencéphale, mais que l’étape initiale de la morphogenèse des bulbes olfactifs est défectueuse. Quant à la défaillance présumée de la migration des cellules à GnRH vers le cerveau, son mécanisme reste assez mal compris, mais on peut supposer qu’elle est secondaire à la désorganisation des fibres du nervus terminalis qui leur servent normalement de support de migration [16-18].
Figure 3
Expression de l’anosmine-1 dans l’ébauche des bulbes olfactifs.
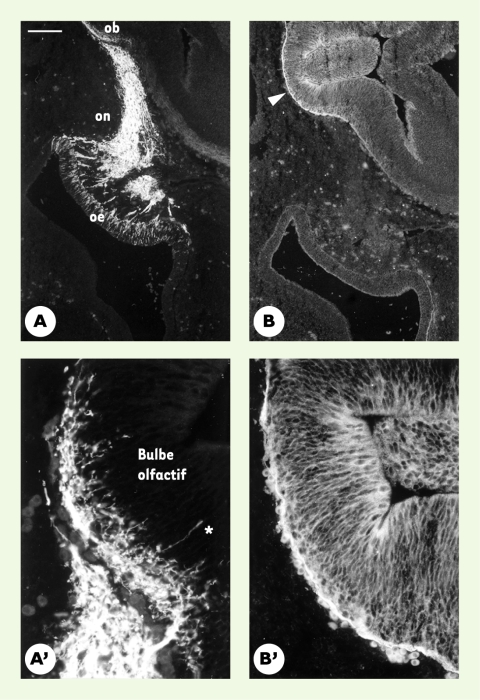
Immunodétection de la protéine NCAM (neural cell adhesion molecule) (A, A’) et de l’anosmine-1 (B, B’) sur des coupes parasagittales adjacentes de la région olfactive d’un foetus humain de six semaines. Les photographies A’ et B’ représentent un agrandissement de la région rostrale du télencéphale correspondant à l’ébauche du bulbe olfactif. Les fibres du nerf olfactif (on), accompagnées par celles du nervus terminalis, sont révélées par leur immunoréactivité NCAM. Les axones des neurones sensoriels olfactifs, dont les corps cellulaires sont situés dans l’épithélium olfactif (oe), sont parvenus jusqu’au cerveau. On peut voir quelques axones pionniers (astérisque) qui pénètrent en profondeur jusqu’à la zone sous-ventriculaire du futur bulbe olfactif (ob). L’anosmine-1 est présente en abondance dans la matrice interstitielle du bulbe olfactif présomptif ainsi que dans la membrane basale qui l’entoure (tête de flèche). En revanche, la protéine n’est détectée ni dans l’épithélium olfactif, ni sur le trajet extracérébral des axones sensoriels. Barre d’échelle : 200 µm en A, B et 50 µm en A’, B’.
Figure 4
Rôle de la signalisation par les FGF dans la morphogenèse des bulbes olfactifs, et mécanisme proposé pour l’aplasie des bulbes olfactifs dans le syndrome de Kallmann De Morsier.

Peu après le premier contact des axones olfactifs avec le télencéphale rostral (au cours de la 6e semaine de développement embryonnaire), les cellules neuro-épithéliales situées au voisinage des terminaisons axonales quittent le cycle mitotique et se différencient en neuroblastes en présence de FGF et de l’anosmine-1, tandis que les cellules situées à distance de ces terminaisons axonales continuent à se multiplier. C’est la diminution régionale de la prolifération cellulaire qui entraînerait l’évagination des bulbes olfactifs primitifs [19]. Cette étape précoce de la morphogenèse des bulbes olfactifs serait défectueuse dans le syndrome de Kallmann De Morsier, notamment par insuffisance quantitative de FGFR1 (KAL-2) ou absence de l’anosmine-1 (KAL-1).
Quelques perspectives de recherche
Bien qu’une mutation particulière de FGFR1 entraînant un « gain de fonction » soit responsable d’une forme de crâniosynostose (i.e. soudure prématurée de certains os du crâne) [20, 21], on pense que les mutations de KAL1 ou de FGFR1 entraînent une perte d’activité des protéines correspondantes [1, 8-10]. Les diverses anomalies cliniques qui peuvent être associées aux mutations de ces gènes (Tableau 1) constituent donc autant de points d’entrée potentiels pour tenter de comprendre le rôle de la signalisation par les FGF dans les phénomènes du développement correspondants. Ainsi peut-on d’ores et déjà expliquer par un déficit de la signalisation via FGFR1 les anomalies de la morphogenèse observées dans la forme KAL-2, telles qu’une fente palatine ou encore des agénésies dentaires [1]. De même, la présence chez certains individus atteints du syndrome de Kallmann De Morsier de syncinésies controlatérales d’imitation (particulièrement fréquentes dans la forme KAL-1), qui pourraient être en rapport avec une anomalie de la décussation du faisceau pyramidal [22], devrait-elle éclairer sur la contribution de la signalisation par les FGF aux processus d’axonogenèse, de fasciculation d’axones et de guidage axonal [23]. Par ailleurs, la participation de l’anosmine-1 au processus de branchement axonal a été montrée récemment dans deux espèces animales, le rat et le nématode Caenorhabditis elegans. Chez le rat, l’application sur un explant de cerveau embryonnaire, d’un anticorps polyclonal dirigé contre l’anosmine-1 humaine inhibe la formation des branches du tractus olfactif latéral [24]. Chez le nématode, la production ectopique et/ou en excès d’anosmine-1 par certains neurones, dans des animaux transgéniques, entraîne un branchement de leurs axones qui n’est pas observé chez les vers normaux [25]. Il reste à déterminer si l’effet stimulant de l’anosmine-1 sur le branchement axonal utilise une signalisation par les FGF. Enfin, si l’étude des souris génétiquement déficientes en Fgfr1 a révélé le rôle essentiel de ce récepteur dans la différenciation des cellules neuro-épithéliales au pôle rostral du télencéphale (bulbe olfactif présomptif) [14], on sait que la signalisation par les FGF participe également à la différenciation des cellules neuro-épithéliales dans d’autres régions du cerveau de l’embryon, en particulier corticales [26, 27]. Le rôle probable de l’anosmine-1 dans ce processus développemental reste à comprendre. La protéine pourrait d’ailleurs aussi être impliquée dans les territoires du cerveau adulte où persiste une neurogenèse (bulbes olfactifs, hippocampe).
Il convient de souligner qu’en termes moléculaires, on ne sait presque rien du rôle de l’anosmine-1. Par son domaine similaire à celui de la protéine acide du petit lait (Figure 2), elle pourrait avoir une activité inhibitrice de protéases à sérine, mais cette hypothèse n’a encore reçu aucune confirmation expérimentale. Quant à ses quatre domaines analogues aux motifs répétés de type III de la fibronectine (Figure 2), on en trouve de semblables dans de nombreuses autres protéines, dont les rôles sont divers. Lorsque l’anosmine-1 est produite par une lignée transfectée de cellules de mammifère, la protéine est détectée principalement au contact de la surface cellulaire. L’interaction de l’anosmine-1 avec la membrane plasmique est dépendante de la présence de glycosaminoglycanes du type héparane sulfate auxquels la protéine se lie [28]. D’ailleurs, le phénotype de branchement axonal que l’on observe chez les vers transgéniques qui surexpriment le gène kal1 dans certains de leurs neurones n’est plus observé chez des animaux mutants déficients en héparane 6O-sulfotransférase, une enzyme impliquée dans la synthèse de ces glycosaminoglycannes [25, 29]. La liaison de l’anosmine-1 aux glycosaminoglycanes constitue, avec le caractère pléïotrope des anomalies cliniques communes aux formes KAL-1 et KAL-2 de la maladie et la colocalisation de l’anosmine-1 avec FGFR1 dans diverses structures de l’embryon, un troisième argument en faveur de l’implication de cette protéine dans la signalisation par les FGF. En effet, les protéoglycanes à héparane sulfate jouent un rôle déterminant dans la dimérisation des récepteurs des FGF en présence de leur ligand (Figure 1), une étape qui conditionne l’activation de la fonction tyrosine kinase de ces récepteurs [30, 31]. Peut-être existe-t-il également une liaison directe de l’anosmine-1 à FGFR1, car on vient de montrer que la protéine NCAM (neural cell adhesion molecule) se lie à la partie extracellulaire de ce récepteur par l’intermédiaire de ses deux modules semblables aux motifs répétés de type III de la fibronectine [32].
Pour finir, rappelons que KAL1 et FGFR1 ne sont impliqués que chez une minorité (environ 20 %) des individus atteints du syndrome de Kallmann De Morsier. Les quelques translocations chromosomiques, toutes différentes, trouvées chez de rares individus atteints de la maladie, indiquent que plusieurs autres gènes sont à découvrir.
Appendices
Note
-
[1]
Région ainsi dénommée en raison de ses propriétés génétiques particulières (absence de liaison au sexe), localisée à l’extrémité du bras court des chromosomes X et Y, commune à ces deux chromosomes. Au cours de la méïose mâle, la région pseudo-autosomique des chromosomes sexuels est le siège d’un appariement et d’un échange de matériel génétique (crossing-over) entre ces deux chromosomes.
Références
- 1. Dodé C, Levilliers J, Dupont J-M, et al.FGFR1 loss-of-function mutations cause autosomal dominant Kallmann syndrome. Nature Genet 2003 ; 33 : 463-5.
- 2. Naftolin F, Harris GW, Bobrow M. Effect of purified luteinizing hormone releasing factor on normal and hypogonadotropic anosmic men. Nature 1971 ; 232 : 496-7.
- 3. De Morsier G. Etudes sur les dysraphies crânio-encéphaliques. 1. Agénésie des lobes olfactifs (telencephaloschizis latéral) et des commissures calleuse et antérieure (telencephaloschizis médian). La dysplasie olfacto-génitale. Schweiz Arch Neurol Psychiat 1954 ; 74 : 309-61.
- 4. Hardelin J-P. Kallmann syndrome : Towards molecular pathogenesis. Mol Cell Endocrinol 2001 ; 179 : 75-81.
- 5. Kallmann FJ, Schoenfeld WA, Barrera SE. The genetic aspects of primary eunuchoidism. Am J Mental Deficiency 1944 ; XLVIII : 203-36.
- 6. Franco B, Guioli S, Pragliola A, et al. A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature 1991 ; 353 : 529-36.
- 7. Legouis R, Hardelin J-P, Levilliers J, et al. The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell 1991 ; 67 : 423-35.
- 8. Ballabio A, Andria G. Deletions and translocations involving the distal short arm of the human X chromosome : Review and hypotheses. Hum Mol Genet 1992 ; 1 : 221-7.
- 9. Hardelin J-P, Levilliers J, Blanchard S, et al. Heterogeneity in the mutations responsible for X chromosome-linked Kallmann syndrome. Hum Mol Genet 1993 ; 2 : 373-7.
- 10. Dodé C, Hardelin J-P. Kallmann syndrome : FGF-signalling insufficiency ? J Mol Med 2004 ; sous presse.
- 11. Oliveira LMB, Seminara SB, Beranova M, et al. The importance of autosomal genes in Kallmann syndrome : Genotype-phenotype correlations and neuroendocrine characteristics. J Clin Endocrinol Metab 2001 ; 86 : 1532-8.
- 12. Hardelin J-P, Julliard AK, Moniot B, et al. Anosmin-1 is a regionally restricted component of basement membranes and interstitial matrices during organogenesis : Implications for the developmental anomalies of X chromosome-linked Kallmann syndrome. Dev Dyn 1999 ; 215 : 26-44.
- 13. Carrel L, Cottle AA, Goglin KC, Willard HF. A first-generation X-inactivation profile of the human X chromosome. Proc Natl Acad Sci USA 1999 ; 96 : 14440-44.
- 14. Hébert JM, Partanen J, Rossant J, McConnell SK. FGF signaling through FGFR1 is required for olfactory bulb morphogenesis. Development 2003 ; 130 : 1101-11.
- 15. Schwanzel-Fukuda M, Pfaff DW. Origin of luteinizing hormone-releasing hormone neurons. Nature 1989 ; 338 : 161-4.
- 16. Schwanzel-Fukuda M, Crossin KL, Pfaff DW, et al. Migration of luteinizing hormone-releasing hormone (LHRH) neurons in early human embryos. J Comp Neurol 1996 ; 366 : 547-57.
- 17. Schwanzel-Fukuda M, Pfaff DW. The structure and function of the nervus terminalis. In : Doty RL, ed. The handbook of clinical olfaction and taste. New York : Dekker, 1995 ; 835-64.
- 18. Schwanzel-Fukuda M, Bick D, Pfaff DW. Luteinizing hormone-releasing hormone (LHRH)-expressing cells do not migrate normally in an inherited hypogonadal (Kallmann) syndrome. Mol Brain Res 1989 ; 6 : 311-26.
- 19. Gong Q, Shipley MT. Evidence that pioneer olfactory axons regulate telencephalon cell cycle kinetics to induce the formation of the olfactory bulb. Neuron 1995 ; 14 : 91-101.
- 20. Muenke M, Schell U, Hehr A, et al. A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nature Genet 1994 ; 8 : 269-74.
- 21. Ibrahimi OA, Zhang F, Eliseenkova AV, et al. Proline to arginine mutations in FGF receptors 1 and 3 result in Pfeiffer and Muenke craniosynostosis syndromes through enhancement of FGF binding affinity. Hum Mol Genet 2004 ; 13 : 69-78.
- 22. Mayston MJ, Harrison LM, Quinton R, et al. Mirror movements in X-linked Kallmann’s syndrome. I. A neurophysiological study. Brain 1997 ; 120 : 1199-216.
- 23. Bülow HE, Boulin T, Hobert O. Differential functions of the C. elegans FGF receptor in axon outgrowth and maintenance of axon position. Neuron 2004 ; 42 : 367-74.
- 24. Soussi-Yanicostas N, de Castro F, Julliard AK, et al. Anosmin-1, defective in the X-linked form of Kallmann syndrome, promotes axonal branch formation from olfactory bulb output neurons. Cell 2002 ; 109 : 217-28.
- 25. Bülow HE, Berry KL, Topper LH, et al. Heparan sulfate proteoglycan-dependent induction of axon branching and axon misrouting by the Kallmann syndrome gene kal-1. Proc Natl Acad Sci USA 2002 ; 99 : 6346-51.
- 26. Raballo R, Rhee J, Lyn-Cook R, et al. Basic fibroblast growth factor (Fgf2) is necessary for cell proliferation and neurogenesis in the developing cerebral cortex. J Neurosci 2000 ; 20 : 5012-23.
- 27. Shin DM, Korada S, Raballo R, et al. Loss of glutamatergic pyramidal neurons in frontal and temporal cortex resulting from attenuation of Fgfr1 signaling is associated with spontaneous hyperactivity in mice. J Neurosci 2004 ; 24 : 2247-58.
- 28. Soussi-Yanicostas N, Hardelin J-P, Arroyo-Jimenez M, et al. Initial characterization of anosmin-1, a putative extracellular matrix protein synthesized by definite neuronal cell populations in the central nervous system. J Cell Sci. 1996 ; 109 : 1749-57.
- 29. Bülow HE, Hobert O. Differential sulfations and epimerization define heparan sulfate specificity in nervous system development. Neuron 2004 ; 41 : 723-36.
- 30. Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2000 ; 103 : 211-25.
- 31. Pellegrini L. Role of heparan sulfate in fibroblast growth factor signaling : a structural view. Curr Opin Struct Biol 2001 ; 11 : 629-34.
- 32. Kiselyov VV, Skladchikova G, Hinsby AM, et al. Structural basis for a direct interaction between FGFR1 and NCAM and evidence for a regulatory role of ATP. Structure 2003 ; 11 : 691-701.
- 33. Schlessinger J. Signal transduction. Autoinhibition control. Science 2003 ; 300 : 750-52.
List of figures
Figure 1
Activation de FGFR1 par ses ligands FGF en présence de protéoglycanes à héparane sulfate.
FGFR1 est un récepteur membranaire actif sous forme de dimère, les monomères étant inactifs. La région extracellulaire, qui fixe le ligand FGF, comporte trois domaines de type immunoglobuline (IgI, IgII et IgIII). Entre les deux premiers s’intercale un module très riche en résidus acides (région acide), dont le rôle serait d’empêcher l’activation spontanée du récepteur en l’absence de son ligand [33]. La région intracellulaire comprend deux domaines à activité tyrosine kinase (TK). La formation d’un complexe moléculaire tripartite entre FGF, FGFR et la chaîne héparane sulfate d’un protéoglycanne est nécessaire à la dimérisation du récepteur, qui est suivie de son activation par autophosphorylation (P) de résidus tyrosine (Y) dans la région intracellulaire de la molécule. Les phosphotyrosines stimulent en retour l’activité tyrosine kinase du récepteur ou servent de points d’ancrage à des molécules de la cascade de signalisation.
Figure 2
Structure de l’anosmine-1.
L’anosmine-1 est une protéine de la matrice extracellulaire. Elle est présente localement, dans divers tissus, pendant la période de l’organogenèse. Son absence est à l’origine de la forme liée au chromosome X du syndrome de Kallmann De Morsier. Elle comporte une région aminoterminale riche en résidus cystéine, au sein de laquelle on identifie un domaine d’environ 50 acides aminés, d’abord mis en évidence dans la protéine acide du petit lait ou whey acidic protein (WAP), et conservé dans toutes les protéines orthologues de l’anosmine-1 humaine connues à ce jour, y compris chez les invertébrés. Ce motif est également présent dans plusieurs petites protéines sécrétées qui ont une activité inhibitrice de protéases à sérine, mais une telle activité demeure hypothétique pour l’anosmine-1 (comme d’ailleurs pour la protéine WAP). Le reste de la protéine comprend quatre domaines semblables aux motifs répétés de type III de la fibronectine, suivis par un court fragment carboxyterminal riche en acides aminés basiques et en histidine. L’anosmine-1 se lie aux glycosaminoglycannes du type héparane sulfate, ce qui suggère qu’elle participe à la signalisation par les facteurs FGF.
Figure 3
Expression de l’anosmine-1 dans l’ébauche des bulbes olfactifs.
Immunodétection de la protéine NCAM (neural cell adhesion molecule) (A, A’) et de l’anosmine-1 (B, B’) sur des coupes parasagittales adjacentes de la région olfactive d’un foetus humain de six semaines. Les photographies A’ et B’ représentent un agrandissement de la région rostrale du télencéphale correspondant à l’ébauche du bulbe olfactif. Les fibres du nerf olfactif (on), accompagnées par celles du nervus terminalis, sont révélées par leur immunoréactivité NCAM. Les axones des neurones sensoriels olfactifs, dont les corps cellulaires sont situés dans l’épithélium olfactif (oe), sont parvenus jusqu’au cerveau. On peut voir quelques axones pionniers (astérisque) qui pénètrent en profondeur jusqu’à la zone sous-ventriculaire du futur bulbe olfactif (ob). L’anosmine-1 est présente en abondance dans la matrice interstitielle du bulbe olfactif présomptif ainsi que dans la membrane basale qui l’entoure (tête de flèche). En revanche, la protéine n’est détectée ni dans l’épithélium olfactif, ni sur le trajet extracérébral des axones sensoriels. Barre d’échelle : 200 µm en A, B et 50 µm en A’, B’.
Figure 4
Rôle de la signalisation par les FGF dans la morphogenèse des bulbes olfactifs, et mécanisme proposé pour l’aplasie des bulbes olfactifs dans le syndrome de Kallmann De Morsier.
Peu après le premier contact des axones olfactifs avec le télencéphale rostral (au cours de la 6e semaine de développement embryonnaire), les cellules neuro-épithéliales situées au voisinage des terminaisons axonales quittent le cycle mitotique et se différencient en neuroblastes en présence de FGF et de l’anosmine-1, tandis que les cellules situées à distance de ces terminaisons axonales continuent à se multiplier. C’est la diminution régionale de la prolifération cellulaire qui entraînerait l’évagination des bulbes olfactifs primitifs [19]. Cette étape précoce de la morphogenèse des bulbes olfactifs serait défectueuse dans le syndrome de Kallmann De Morsier, notamment par insuffisance quantitative de FGFR1 (KAL-2) ou absence de l’anosmine-1 (KAL-1).
List of tables
Tableau I
Formes génétiques identifiées du syndrome de Kallmann De Morsier.
FGF : fibroblast growth factors ; FGFR1 : fibroblast growth factor receptor 1.