Abstracts
Résumé
En raison de sa capacité à réagir avec l’oxygène et à changer d’état d’oxydoréduction, le fer est indispensable à toute forme de vie sur terre. La découverte récente de nombreuses enzymes à activité ferroxydase (héphaestine, céruloplasmine) ou ferriréductase (Dcytb), ainsi que de transporteurs de cations divalents (Nramp2/DMT1, ferroportine) impliqués dans le transport du fer a mis en évidence l’existence d’un réseau protéique complexe et finement réglé permettant de mobiliser le fer à partir des aliments, de le transporter à travers les membranes cellulaires et de le véhiculer dans les fluides biologiques. L’homéostasie du fer repose sur un contrôle strict de son absorption intestinale, au niveau du duodénum, et de son recyclage après dégradation des globules rouges sénescents par les macrophages. L’hepcidine, un peptide synthétisé par le foie et contrôlé par le fer, ainsi que la protéine HFE, dont les mutations sont responsables de la forme prédominante d’hémochromatose héréditaire, sont impliquées dans ces régulations. Ces découvertes ouvrent de nouvelles perspectives thérapeutiques, notamment en ce qui concerne la possibilité de traiter les surcharges en fer par l’hepcidine.
Summary
Iron metabolism in mammals requires a complex and tightly regulated molecular network. The classical view of iron metabolism has been challenged over the past ten years by the discovery of several new proteins, mostly Fe (II) iron transporters, enzymes with ferro-oxydase (hephaestin or ceruloplasmin) or ferri-reductase (Dcytb) activity or regulatory proteins like HFE and hepcidin. Furthermore, a new transferrin receptor has been identified, mostly expressed in the liver, and the ability of the megalin-cubilin complex to internalise the urinary Fe (III)-transferrin complex in renal tubular cells has been highlighted. Intestinal iron absorption by mature duodenal enterocytes requires Fe (III) iron reduction by Dcytb and Fe (II) iron transport through apical membranes by the iron transporter Nramp2/DMT1. This is followed by iron transfer to the baso-lateral side, export by ferroportin and oxidation into Fe (III) by hephaestin prior to binding to plasma transferrin. Macrophages play also an important role in iron delivery to plasma transferrin through phagocytosis of senescent red blood cell, heme catabolism and recycling of iron. Iron egress from macrophages is probably also mediated by ferroportin and patients with heterozygous ferroportin mutations develop progressive iron overload in liver macrophages. Iron homeostasis at the level of the organism is based on a tight control of intestinal iron absorption and efficient recycling of iron by macrophages. Signalling between iron stores in the liver and both duodenal enterocytes and macrophages is mediated by hepcidin, a circulating peptide synthesized by the liver and secreted into the plasma. Hepcidin expression is stimulated in response to iron overload or inflammation, and down regulated by anemia and hypoxia. Hepcidin deficiency leads to iron overload and hepcidin overexpression to anemia. Hepcidin synthesis in response to iron overload seems to be controlled by the HFE molecule. Patients with hereditary hemochromatosis due to HFE mutation have impaired hepcidin synthesis and forced expression of an hepcidin transgene in HFE deficient mice prevents iron overload. These results open new therapeutic perspectives, especially with the possibility to use hepcidin or antagonists for the treatment of iron overload disorders.
Article body
Le fer est un élément paradoxal, indispensable à toute forme de vie, essentiellement pour assurer le transport d’oxygène et catalyser des réactions de transfert d’électrons, de fixation d’azote ou de synthèse d’ADN, mais également toxique, en raison de sa capacité à réagir avec l’oxygène et à catalyser la production de formes radicalaires.
Les organismes vivants ont développé un grand nombre de protéines permettant de véhiculer le fer dans les fluides biologiques ou à travers les membranes cellulaires et de le mettre en réserve sous une forme facilement disponible, mais non toxique. À l’inverse de la ferritine, protéine de stockage du fer très conservée, les systèmes d’acquisition du fer ont considérablement évolué entre les espèces et se sont adaptés à la forme chimique sous laquelle le fer se trouve dans l’environnement [1].
Chez les mammifères, le fer circulant plasmatique est lié à la transferrine. L’internalisation du complexe fer-transferrine par l’intermédiaire de récepteurs spécifiques, présents en grand nombre à la surface des cellules, a longtemps été considérée comme la voie principale d’acquisition du fer par les cellules [2]. Cependant, l’étude d’animaux génétiquement modifiés ou porteurs de mutations spontanées a permis d’identifier de nouvelles protéines impliquées dans le transport du fer à travers les membranes biologiques, en particulier des transporteurs du Fe ferreux (II). De plus, une deuxième forme du récepteur de la transferrine, principalement exprimé dans le foie, a récemment été mise en évidence [3], de même qu’une autre voie d’internalisation de la transferrine à partir du complexe mégaline-cubiline [4] ((→) m/s 2003, n°5, p.337).
Les systèmes d’acquisition du fer contrôlent la charge en fer de l’organisme
Il n’existe pas de moyen pour l’organisme d’éliminer le fer absorbé en excès, et la constitution d’une surcharge en fer de l’organisme ne peut être évitée que par un contrôle fin de l’absorption intestinale (Figure 1A) et du recyclage macrophagique (Figure 1B). Les mécanismes de régulation de l’absorption intestinale du fer sont restés longtemps méconnus, mais des progrès notables ont été accomplis par l’identification du gène responsable de l’hémochromatose génétique, codant pour la protéine HFE, et dernièrement par la découverte de l’hepcidine, un peptide circulant jouant un rôle majeur dans l’homéostasie du fer.
Figure 1
Acquisition du fer par l’entérocyte duodénal et le macrophage.
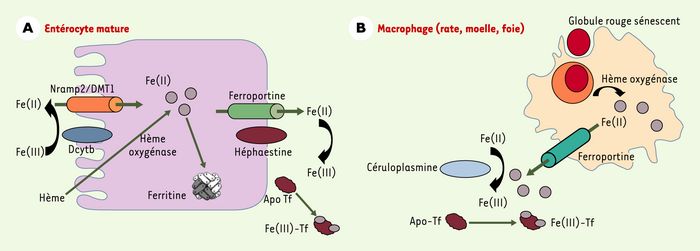
A. Les entérocytes matures, au sommet de la villosité duodénale, absorbent le fer à partir des aliments et assurent son transfert vers le plasma. Au pôle apical, Dcytb, une réductase membranaire, réduit le Fe (III) en Fe (II) [5], celui-ci étant par la suite transporté à travers la membrane par Nramp2/DMT1 [6-8]. Un transport vectoriel du fer cytoplasmique, par un mécanisme encore inconnu, permet au fer d’être ciblé vers le pôle basolatéral et exporté vers le plasma par la ferroportine. Le fer est ensuite réoxydé par l’héphaestine [12] et pris en charge par la transferrine plasmatique (Apo Tf). Le fer non transféré au plasma est stocké par la ferritine, puis éliminé par désquamation des entérocytes matures. Le fer héminique absorbé au pôle apical sera libéré par l’hème oxygénase et exporté de la même façon que le fer non héminique. B. L’organisme d’un être humain adulte contient environ 4 g de fer, dont plus de la moitié est associée à l’hémoglobine des érythrocytes circulants. La phagocytose des globules rouges sénescents par les macrophages assure un recyclage efficace des atomes de fer vers le plasma [13]: environ 25 mg de fer pourraient ainsi être recyclés quotidiennement pour participer à l’érythropoïèse. Ce mécanisme concerne principalement les macrophages de la rate et de la moelle osseuse et, dans une moindre mesure, les cellules de Kupffer. Le fer libéré par le catabolisme de l’hème assuré par l’hème oxygénase peut être stocké par la molécule de ferritine ou recyclé vers le plasma. Les mécanismes de recyclage sont encore mal connus, mais la protéine ferroportine/IREG1/MTP1, une protéine transmembranaire qui exporte le Fe (II), semble jouer un rôle important dans ce processus [9-11, 14, 15]. Le fer ferreux est ensuite oxydé par la céruloplasmine sérique [16] et fixé par la transferrine. Il est probable que la céruloplasmine est également impliquée dans les échanges de fer entre plusieurs tissus, des malades porteurs d’une acéruloplasminémie héréditaire développant progressivement une surcharge en fer qui s’accompagne d’un diabète, d’une dégénérescence rétinienne ou de symptômes neurologiques [17].
Un modèle de régulation de l’absorption intestinale a été proposé, qui suggère que les cellules indifférenciées de la crypte reçoivent des signaux émis par l’organisme, leur permettant d’exprimer, au cours de leur différenciation et de leur migration le long de la villosité intestinale, les protéines nécessaires à l’absorption du fer, à un niveau adapté aux besoins de l’organisme. Ces signaux, de deux types, permettraient de moduler l’absorption en fonction du niveau des réserves en fer (store regulator) ou de l’activité érythropoïétique de la moelle osseuse (erythroid regulator) [18]. La nature moléculaire de ces signaux est encore incomplètement élucidée, mais il est sûr que les protéines HFE et hepcidine jouent un rôle majeur dans cette signalisation.
Intervention de la protéine HFE
La protéine HFE, dont une mutation est responsable de l’hémochromatose génétique, est une molécule HLA de classe I non classique [19]. Des expériences de cocristallisation et de co-immunoprécipitation ont clairement démontré que la protéine HFE s’associe avec le récepteur de la transferrine [20], facilitant ainsi l’endocytose du complexe fer-transferrine et modulant la quantité de fer reçue par les cellules. La mutation Cys282Tyr de la protéine HFE, retrouvée chez la majorité des patients atteints d’hémochromatose héréditaire, empêche son ciblage à la membrane et conduit à sa dégradation rapide. L’absence d’HFE fonctionnelle diminuerait la quantité de fer reçue par les cellules, en particulier par les cellules indifférenciées de la crypte duodénale, qui percevraient cette situation comme un signal de carence en fer et continueraient à exprimer les protéines de transport du fer, malgré des réserves en fer tissulaires supérieures à la normale (Figure 2). Ce modèle a cependant été remis en question par la découverte récente de l’hepcidine, un régulateur majeur de l’homéostasie du fer, et par la mise en évidence du rôle de la protéine HFE dans l’expression de l’hepcidine.
Figure 2
Régulation de l’homéostasie du fer.
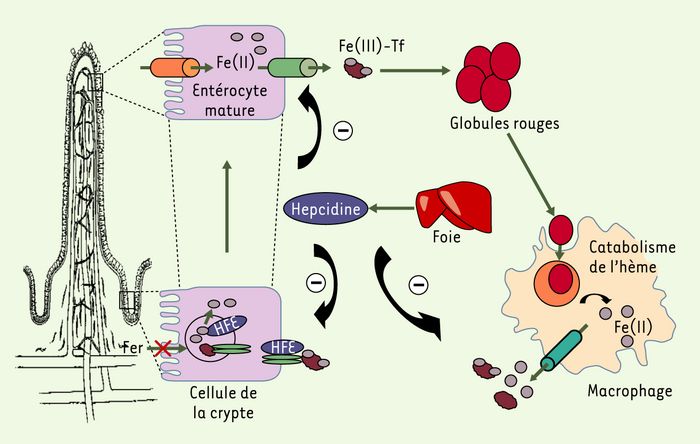
Au cours de leur migration le long la villosité duodénale, les cellules de la crypte se différencient et expriment les protéines de capture et de transfert du fer, à un niveau variable selon les besoins en fer de l’organisme. Cette programmation dépend de signaux, émis par l’organisme, dont la nature moléculaire est encore imprécise. La molécule HFE, exprimée au pôle basolatéral des cellules de la crypte, interagit avec les récepteurs de la transferrine (ovales verts) et contrôle la quantité de fer internalisé. Lorsque la protéine est mutée, comme c’est le cas dans l’hémochromatose génétique, le duodénum perçoit un signal de carence en fer et continue à exprimer les protéines de l’absorption malgré des réserves en fer élevées. Un autre modèle propose qu’en l’absence d’HFE fonctionnelle, l’activation de la synthèse d’hepcidine après l’augmentation des réserves en fer ne se fasse plus normalement, entraînant le développement d’une surcharge. En effet, l’hepcidine, synthétisée par le foie et sécrétée dans le plasma, contrôle négativement aussi bien l’absorption intestinale du fer que le recyclage macrophagique du fer héminique, libéré par la phagocytose des globules rouges sénescents et le catabolisme de l’hème.
Rôle de l’hepcidine
L’hepcidine est synthétisée par le foie sous forme d’un précurseur de 80 acides aminés, puis sécrétée dans le plasma sous forme d’un peptide mature de 20-25 acides aminés, très structuré par la présence de 8 cystéines formant 4 ponts disulfure [21]. L’hepcidine, isolée des urines comme peptide antimicrobien, a ensuite été identifiée comme une protéine hépatique dont la synthèse est fortement inductible, in vivo comme in vitro, par une surcharge en fer et par les lipopolysaccharides bactériens [22]. La confirmation de son rôle dans le contrôle de l’homéostasie du fer est venue de l’étude de souris recombinantes pour le gène codant pour le facteur de transcription USF2 (upstream stimulatory factor), qui présentaient accidentellement une inactivation du gène codant pour l’hepcidine [23]. Le défaut d’expression d’hepcidine entraîne chez les animaux une surcharge en fer progressive du foie et du pancréas, ainsi qu’une déplétion des réserves en fer des macrophages tissulaires. L’hepcidine circulante pourrait être le store regulator recherché depuis plusieurs années, contrôlant la sortie du fer des entérocytes duodénaux et des macrophages.
Le mode d’action de l’hepcidine est encore inconnu, mais la protéine pourrait avoir un effet direct, sur l’activité d’exportation du fer, ou indirect, en modulant le niveau d’expression des protéines de transport. L’importance de l’hepcidine chez l’homme a été démontrée par la mise en évidence de mutations inactivatrices de ce peptide chez deux familles atteintes d’hémochromatose juvénile [24]. Par ailleurs, le passage transplacentaire du fer semble pouvoir être inhibé par l’hepcidine, puisque des souris transgéniques surexprimant l’hepcidine présentent une anémie néonatale sévère et meurent quelques heures après la naissance [25].
La similarité phénotypique (surcharge hépatique en fer et diminution du contenu en fer des macrophages) des souris déficientes en protéine HFE ou déficientes en hepcidine a dès le début suggéré que les deux protéines intervenaient sur une même voie de transduction du signal entre le fer tissulaire et l’entérocyte. Cette hypothèse a été renforcée par la mise en évidence, chez la souris [26] comme chez l’homme [27], d’un défaut d’activation de la synthèse d’hepcidine en réponse à une surcharge en fer lorsque la protéine HFE est mutée ou inactivée. De plus, l’introduction d’un transgène hepcidine chez des souris déficientes en HFE permet d’empêcher l’apparition d’une surcharge en fer [26]. L’ensemble de ces résultats suggère que la protéine HFE participe à la régulation de l’expression de l’hepcidine, en fonction de la charge en fer de l’organisme.
Des modèles murins, ainsi que des dosages d’hepcidine urinaire chez l’homme, ont montré que les états inflammatoires ou infectieux stimulent très fortement l’expression de l’hepcidine [28, 29], suggérant que ce médiateur soluble pourrait également contribuer aux anémies fréquemment associées à ces situations pathologiques. Enfin, la répression de l’hepcidine observée au cours d’une anémie hémolytique expérimentale ou en situation d’hypoxie [28] est probablement une condition nécessaire, mais pas nécessairement suffisante, à la stimulation de l’absorption intestinale du fer après activation de l’érythropoïèse. Il est intéressant de noter que les érythropoïèses inefficaces avec hémolyse intramédullaire stimulent très fortement l’absorption intestinale du fer, contrairement aux hémolyses périphériques [18], par un mécanisme qui demeure inconnu.
Ces différents résultats ouvrent des perspectives intéressantes pour l’utilisation de l’hepcidine comme agent thérapeutique dans le traitement des surcharges en fer acquises, secondaires à des anomalies de l’érythropoïèse comme celles observées dans les thalassémies, et peut être aussi dans certaines formes héréditaires d’hémochromatose.
Conclusions
La connaissance du métabolisme du fer a considérablement évolué au cours de cette dernière décennie. D’une vision simplifiée, fondée sur un mode unique aussi bien d’acquisition du fer par les cellules via les récepteurs de la transferrine que de stockage lié à la ferritine, elle est passée à une vision complexe, faisant apparaître une spécialisation importante des différents types cellulaires dans les mécanismes d’acquisition du fer comme dans les régulations coordonnées des différents gènes impliqués. L’identification de nouveaux éléments majeurs de régulation de l’homéostasie du fer, en particulier d’une molécule HLA de classe I - la protéine HFE - et d’un peptide à activité antimicrobienne - l’hepcidine -, ainsi que la découverte d’enzymes à activité ferroxydase ou ferriréductase et de transporteurs membranaires du fer ferreux, ont marqué ces dernières années et fait de l’étude du métabolisme du fer un domaine passionnant. De nouvelles maladies génétiques ont été découvertes, liées à des mutations de l’un ou l’autre des gènes impliqués, et le fer s’est révélé jouer un rôle aggravant dans des situations pathologiques diverses - états inflammatoires, infections, pathologies cardiovasculaires, diabète ou encore insuffisance rénale. De nouvelles perspectives thérapeutiques s’ouvrent, notamment avec l’hepcidine, permettant d’espérer une amélioration des traitements supplétifs en fer, toujours très imparfaits, et des traitements des surcharges en fer, actuellement fondés sur l’utilisation de chélateurs, d’un maniement souvent contraignant pour le malade et non toujours dénués d’effets secondaires.
Appendices
Références
- 1. Nelson N. Metal ion transporters and homeostasis. EMBO J 1999; 18: 4361-71.
- 2. Ponka P, Beaumont C, Richardson DR. Function and regulation of transferrin and ferritin. Semin Hematol 1998; 35: 35-54.
- 3. Trinder D, Baker E. Transferin receptor 2: a new molecule in iron metabolism. Int J Biochem Cell Biol 2003; 35: 292-6.
- 4. Verroust PJ, Kozyraki R. Cubiline: rôle physiopathologique et relations avec la mégaline. Med Sci(Paris) 2003; 19: 337-43.
- 5. McKie AT, Barrow D, Latunde-Dada GO, et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 2001; 291: 1755-9.
- 6. Gunshin H, Mackenzie B, Berger UV, et al. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997; 388: 482-8.
- 7. Canonne-Hergaux F, Gruenheid S, Ponka P, Gros P. Cellular and subcellular localization of the Nramp2 iron transporter in the intestinal brush border and regulation by dietary iron. Blood 1999; 93: 4406-17.
- 8. Fleming MD, Trenor CC III, Su MA, et al. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet 1997; 16: 383-6.
- 9. McKie AT, Marciani P, Rolfs A, et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell 2000; 5: 299-309.
- 10. Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem 2000; 275: 19906-12.
- 11. Donovan A, Brownlie A, Zhou Y, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000; 403: 776-81.
- 12. Vulpe CD, Kuo YM, Murphy TL, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 1999; 21: 195-9.
- 13. Knutson M, Wessling-Resnick M. Iron metabolism in the reticuloendothelial system. Crit Rev Biochem Biol 2003; 38: 61-88.
- 14. Montosi G, Donovan A, Totaro A, et al. Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J Clin Invest 2001; 108: 619-23.
- 15. Devalia V, Carter K, Walker AP, et al. Autosomal dominant reticuloendothelial iron overload associated with a 3-base pair deletion in the ferroportin 1 gene (SLC11A3). Blood 2002; 100: 695-7.
- 16. Harris ZL, Durley AP, Man TK, Gitlin JD. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc Natl Acad Sci USA 1999; 96: 10812-7.
- 17. Harris ZL, Klomp LWJ, Gitlin JD. Aceruloplasminemia: an inherited neurodegenerative disease with impairment of iron homeostasis. Am J Clin Nutr 1998; 67: 972S.
- 18. Andrews NC. Disorders of iron metabolism. N Engl J Med 1999; 341: 1986-95.
- 19. Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 1996; 13: 399-408.
- 20. Ramalingam TS, West AP Jr, Lebron JA, et al. Binding to the transferrin receptor is required for endocytosis of HFE and regulation of iron homeostasis. Nat Cell Biol 2000; 2: 953-7.
- 21. Hunter HN, Fulton DB, Ganz T, Vogel HJ. The solution structure of human hepcidin, a peptide hormone with antimicrobial activity that is involved in iron uptake and hereditary hemochromatosis. J Biol Chem 2002; 277: 37597-603.
- 22. Pigeon C, Ilyin G, Courselaud B, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem 2001; 276: 7811-9.
- 23. Nicolas G, Bennoun M, Devaux I, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA 2001; 98: 8780-5.
- 24. Roetto A, Papanikolaou G, Politou M, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet 2003; 33: 21-2.
- 25. Nicolas G, Bennoun M, Porteu A, et al. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc Natl Acad Sci USA 2002; 99: 4596-601.
- 26. Nicolas G, Viatte L, Lou DQ, et al. Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis. Nat Genet 2003; 34: 97-101.
- 27. Bridle KR, Frazer DM, Wilkins SJ, et al. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet 2003; 361: 669-73.
- 28. Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 2003; 101: 2461-3.
- 29. Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 2002; 110: 1037-44.
List of figures
Figure 1
Acquisition du fer par l’entérocyte duodénal et le macrophage.
A. Les entérocytes matures, au sommet de la villosité duodénale, absorbent le fer à partir des aliments et assurent son transfert vers le plasma. Au pôle apical, Dcytb, une réductase membranaire, réduit le Fe (III) en Fe (II) [5], celui-ci étant par la suite transporté à travers la membrane par Nramp2/DMT1 [6-8]. Un transport vectoriel du fer cytoplasmique, par un mécanisme encore inconnu, permet au fer d’être ciblé vers le pôle basolatéral et exporté vers le plasma par la ferroportine. Le fer est ensuite réoxydé par l’héphaestine [12] et pris en charge par la transferrine plasmatique (Apo Tf). Le fer non transféré au plasma est stocké par la ferritine, puis éliminé par désquamation des entérocytes matures. Le fer héminique absorbé au pôle apical sera libéré par l’hème oxygénase et exporté de la même façon que le fer non héminique. B. L’organisme d’un être humain adulte contient environ 4 g de fer, dont plus de la moitié est associée à l’hémoglobine des érythrocytes circulants. La phagocytose des globules rouges sénescents par les macrophages assure un recyclage efficace des atomes de fer vers le plasma [13]: environ 25 mg de fer pourraient ainsi être recyclés quotidiennement pour participer à l’érythropoïèse. Ce mécanisme concerne principalement les macrophages de la rate et de la moelle osseuse et, dans une moindre mesure, les cellules de Kupffer. Le fer libéré par le catabolisme de l’hème assuré par l’hème oxygénase peut être stocké par la molécule de ferritine ou recyclé vers le plasma. Les mécanismes de recyclage sont encore mal connus, mais la protéine ferroportine/IREG1/MTP1, une protéine transmembranaire qui exporte le Fe (II), semble jouer un rôle important dans ce processus [9-11, 14, 15]. Le fer ferreux est ensuite oxydé par la céruloplasmine sérique [16] et fixé par la transferrine. Il est probable que la céruloplasmine est également impliquée dans les échanges de fer entre plusieurs tissus, des malades porteurs d’une acéruloplasminémie héréditaire développant progressivement une surcharge en fer qui s’accompagne d’un diabète, d’une dégénérescence rétinienne ou de symptômes neurologiques [17].
Figure 2
Régulation de l’homéostasie du fer.
Au cours de leur migration le long la villosité duodénale, les cellules de la crypte se différencient et expriment les protéines de capture et de transfert du fer, à un niveau variable selon les besoins en fer de l’organisme. Cette programmation dépend de signaux, émis par l’organisme, dont la nature moléculaire est encore imprécise. La molécule HFE, exprimée au pôle basolatéral des cellules de la crypte, interagit avec les récepteurs de la transferrine (ovales verts) et contrôle la quantité de fer internalisé. Lorsque la protéine est mutée, comme c’est le cas dans l’hémochromatose génétique, le duodénum perçoit un signal de carence en fer et continue à exprimer les protéines de l’absorption malgré des réserves en fer élevées. Un autre modèle propose qu’en l’absence d’HFE fonctionnelle, l’activation de la synthèse d’hepcidine après l’augmentation des réserves en fer ne se fasse plus normalement, entraînant le développement d’une surcharge. En effet, l’hepcidine, synthétisée par le foie et sécrétée dans le plasma, contrôle négativement aussi bien l’absorption intestinale du fer que le recyclage macrophagique du fer héminique, libéré par la phagocytose des globules rouges sénescents et le catabolisme de l’hème.