Abstracts
Résumé
La perte de fonction β-cellulaire joue un rôle central dans la physiopathologie des diabètes de type 2. Alors que dans les formes rares de la maladie, les MODY (maturity onset diabetes of the young), son étiologie est simple, monogénique et décryptée, dans les formes courantes de diabète de type 2, elle est certainement beaucoup plus complexe et demeure mystérieuse. Cet article établit le bilan des données actuellement disponibles relatives au déterminisme de l’atteinte β-cellulaire dans un modèle animal approprié, le rat GK, spontanément porteur d’un diabète de type 2.
Summary
The pathways that control insulin release and regulate pancreatic β-cell mass are crucial on the development of type 2 diabetes mellitus. Maturity-onset diabetes of the young comprises a number of single-gene disorders affecting β-cell development and/or function. A genetic basis for the more common forms of type 2 diabetes which affect adults in developed as well as many developing countries is less clear cut. It is also characterized by abnormal β-cell function. Appropriate inbred rodent models are an essential tool for the identification of genes and environmental factors that increase the risk of type 2 diabetes. The informations available from studies in the Goto-Kakizaki (GK) rat are here reviewed in such a perspective. This model was obtained by selective breeding of individuals with mild glucose intolerance from a non-diabetic Wistar rat colony. Heritability of defective β-mass and β-cell function in GK model is proposed to reflect the complex interactions of three pathogenic players: (1) three independent loci containing genes causating impaired insulin secretion; (2) gestational metabolic (hyperglycaemic) impairment inducing a programming of endocrine pancreas (decreased β-cell mass) which is transmitted to the next generation; (3) secondary (acquired) loss of β-cell differentiation due to chronic exposure to hyperglycaemia (glucotoxicity). A better understanding of the mechanisms involved in the failure of β-cell function in the GK model will lead to identification of new therapeutic targets for both the prevention and treatment of type 2 diabetes.
Article body
Le diabète sucré n’est pas une maladie homogène. Il s’agit en fait d’un ensemble hétérogène de désordres métaboliques qui, néanmoins, ont comme caractéristique commune de permettre l’installation d’une hyperglycémie chronique. La cellule β pancréatique et son produit de sécrétion, l’insuline, jouent un rôle central dans la physiopathologie des diabètes. Le diabète de type 1, ou diabète insulinodépendant, résulte d’un déficit primaire, en général absolu, en insuline, dû à la destruction auto-immune des cellules β.
Au cours du diabète de type 2 (ou diabète non insulinodépendant), les muscles squelettiques et les tissus adipeux blancs sont devenus résistants à l’action biologique de l’insuline. Par ailleurs, l’adaptation compensatrice des cellules β visant à produire et à libérer chroniquement plus d’insuline dans la circulation (l’hyperinsulinisme ayant pour fonction de compenser l’insulinorésistance) n’est plus suffisante pour assurer la normoglycémie (en particulier en phase post-prandiale), ce qui se traduit in fine par un épuisement fonctionnel des cellules β survivantes [1]. Le diabète de type 2 lui-même n’est pas une maladie homogène, les diverses variantes de diabète type 2 correspondant à un éventail de combinaisons, en proportions variables, des deux traits phénotypiques majeurs, l’insulinorésistance et le déficit de production d’insuline. À l’une des extrémités de cet éventail, on trouve des variantes dont le déterminisme, simple et monogénique, touche la capacité des cellules β de sécréter de l’insuline: ce sont les MODY (maturity onset diabetes of the young) ((→)m/s 1998, n° 3, p.364).
Les bases génétiques de la pathologie β -cellulaire dans les formes humaines
Les MODY représentent des variantes rares de la maladie, dans lesquelles le diabète n’est jamais cétonique; elles se déclarent en général avant 25 ans et leur mode de transmission est autosomique dominant. L’anomalie primaire affecte toujours le fonctionnement de la cellule β. Un MODY peut résulter de mutations de l’un parmi au moins six gènes différents qui codent soit pour une enzyme de la glycolyse, la glucokinase, soit pour l’un des cinq facteurs de transcription suivants: HNF4-α, HNF1-α, Pdx1, HNF1-β et NeuroD1/β2. Tous ces gènes sont exprimés dans la cellule β normale et leurs mutations entraînent des anomalies fonctionnelles des cellules β chez les sujets porteurs hétérozygotes ((→)m/s 2003, n°8-9, p.???).
Indépendamment des mutations touchant les gènes précités qui appartiennent au génome nucléaire, des mutations du génome mitochondrial, rares elles aussi, sont sources d’anomalies de fonctionnement des mitochondries et peuvent également entraîner l’apparition d’un diabète [2].
L’étiologie de la perte de fonction des cellules β dans les formes courantes de diabète de type 2 est certainement beaucoup plus complexe que dans les formes rares. Chez les sujets porteurs d’un diabète de type 2, les anomalies de la sécrétion d’insuline coexistent le plus souvent avec une réduction de lbiologique de l’insuline sur les tissus cibles (insulinorésistance) ((→)m/s 1999, n°8-9, p. 1060).
Mais l’apparition de l’hyperglycémie est due, là encore, à l’effondrement progressif de la production d’insuline résultant de l’incapacité des cellules β de s’adapter au long cours à l’insulinorésistance.
L’existence d’une ségrégation familiale du diabète de type 2, et d’une concordance pour cette maladie beaucoup plus élevée chez les vrais jumeaux que chez les faux jumeaux, suggère un rôle important de l’héritabilité dans l’étiologie de ce diabète. Il existe également de nombreuses études épidémiologiques suggérant qu’un génotype particulier, dit «de l’épargnant» (thrifty genotype), puisse contribuer à l’augmentation de la prévalence des diabètes de type 2, augmentation retrouvée le plus souvent dans les populations humaines ayant changé brutalement leurs habitudes alimentaires et leur activité physique au cours du xxesiècle [3].
Quels sont les gènes responsables du génotype de l’épargnant? Il faut bien reconnaître qu’après plusieurs années d’efforts intensifs et coûteux, l’entreprise d’identification chez l’homme des gènes de susceptibilité au diabète de type 2 n’a pas abouti.
Le paradigme du rat Goto-Kakizaki (GK) ou les attraits d’un modèle animal de diabète spontané de type 2
La plupart des modèles animaux (souris db/db, ob/ob, rat ZDF - Zucker diabetic fatty, rat OLETF - Otsuka Long-Evans Tokushima fatty) actuellement utilisés dans les études de transmission du diabète de type 2 sont également des modèles d’obésité ((→)m/s 1999, n°10, p. 1187). Dans la lignée consanguine Goto-Kakisaki (GK), les animaux adultes, quel que soit leur sexe, n’ont pas de surpoids et présentent une hyperglycémie à jeun modérée et stable (> 18 mois). La lignée GK a été établie à partir d’une colonie de rats Wistar normoglycémiques, par des croisements consanguins répétés des quelques individus sélectionnés à chaque génération sur la base d’une tolérance glucidique infra-normale, évaluée lors d’un test standardisé d’hyperglycémie provoquée par voie orale (Figure 1) [4]. Chez les rats GK élevés dans notre laboratoire (Paris, France) (sous-lignée GK/Par) [5], l’hyperglycémie diabétique, lorsqu’elle est installée, est accompagnée de la triade pathologique caractéristique du diabète de type 2 humain: hyperproduction hépatique de glucose, insulinorésistance périphérique (et ceci en l’absence d’une obésité avérée) et déficit de l’insulinosécrétion (dû à la conjonction d’un déficit numérique de cellules β et d’une absence de réactivité des cellules β résiduelles au glucose). Avant l’installation de l’hyperglycémie, qui se fait au moment du sevrage, entre 3 et 4 semaines, il existe une période de normoglycémie chez tous les jeunes rats GK, qui correspond à une phase de pré-diabète bien identifiable (Figure 2) [5]. La Figure 3 fait le point sur l’état actuel de nos connaissances des anomalies fonctionnelles intrinsèques à la cellule β chez le rat GK diabétique.
Figure 1
Le rat GK (désigné par les initiales du nom des chercheurs Goto et Kakizaki ayant établi cette lignée) est un modèle de diabète spontané de type 2 sans obésité associée.
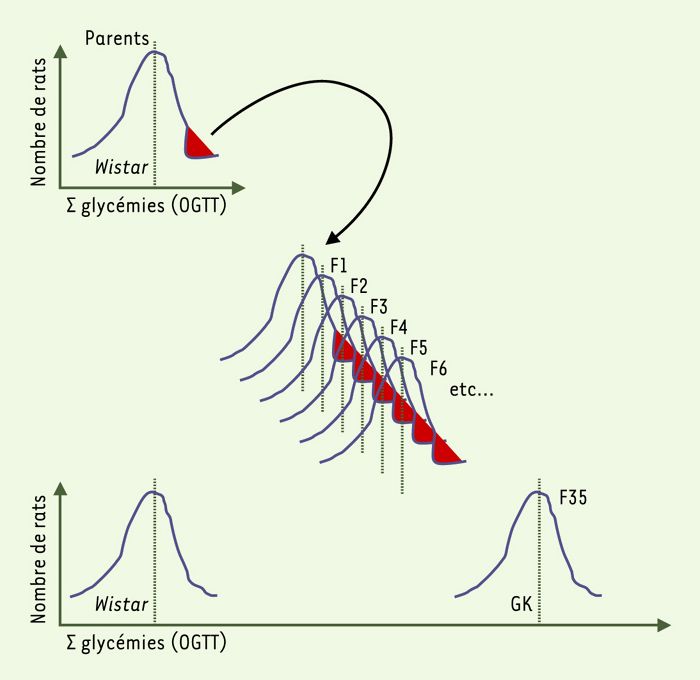
Cette lignée de rat a été obtenue par des croisements sélectifs de rats normaux, de souche Wistar, sélectionnés sur la base d’une légère intolérance au glucose. Cette dernière a été quantifiée par la somme (Σ glycémies) des valeurs de glycémies lors d’un test de surcharge glucosée administrée par voie orale (OGTT). La répétition de cette procédure sur de multiples générations (plus de 30) a permis d’obtenir un état diabétique stable [4]. Les animaux de la sous-lignée GK/Par sont les descendants de rats GK appartenant à la F35 de la lignée japonaise originelle [5].
Figure 2
Chronologie d’apparition et d’évolution du diabète spontané du rat GK.
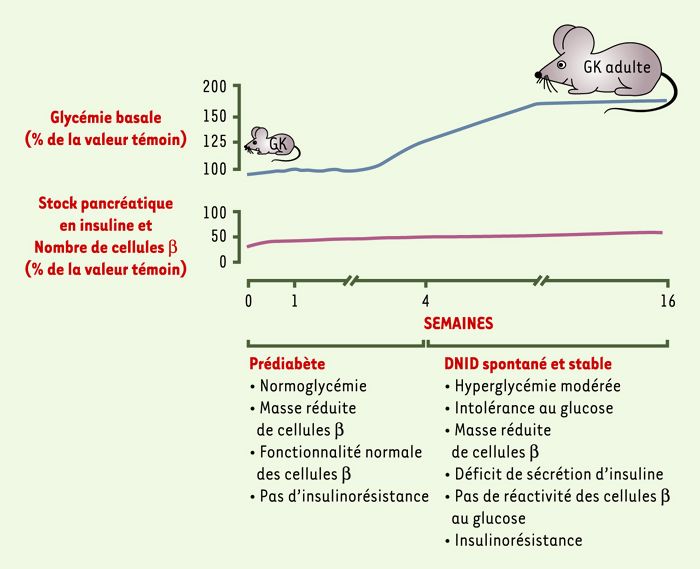
DNID: diabète non insulinodépendant.
Figure 3
Voies de signalisation altérées dans la cellule β du rat GK diabétique.
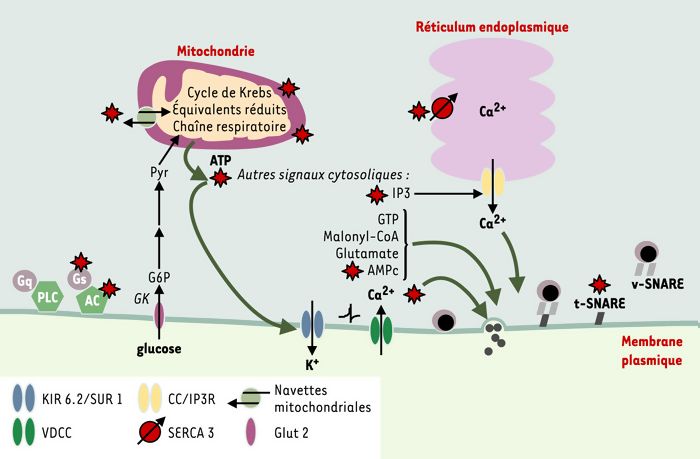
Les anomalies spécifiques de la cellule β GK sont indiquées par le symbole «étoile». Le glucose est phosphorylé (G6P) par la glucokinase (GK) et converti en pyruvate (Pyr) au cours de la glycolyse. Le pyruvate entre préférentiellement dans les mitochondries et alimente la voie des acides tricarboxyliques (cycle de Krebs). Il en résulte un transfert accru d’équivalents réduits à la chaîne respiratoire, ce qui crée les conditions d’une hyperpolarisation de la membrane interne mitochondriale et d’une synthèse accrue d’ATP. C’est la fermeture ultérieure des canaux K+ dépendant de l’ATP (KIR 6.2//SUR 1) qui dépolarise la membrane plasmique. Cela va provoquer l’ouverture de canaux membranaires Ca2+ de type L dépendant du potentiel (VDCC), à l’origine de l’élévation brusque de la concentration cytosolique Ca2+ qui, in fine, activera l’exocytose des vésicules d’insuline (d’après [5]). Il existe également toute une série de signaux cytosoliques additionnels qui participent au couplage métabolisme intracellulaire-sécrétion: ce sont des composés phosphorylés, nucléotidiques ou non (ATP, GTP, AMPc, IP3), ou des métabolites (malonyl-CoA/acyl-CoA, glutamate). Ces signaux contribuent globalement à amplifier le mécanisme déclenchant décrit ci-dessus et interviennent dans des étapes plutôt distales de l’exocytose: ils modulent le fonctionnement du cytosquelette et l’accrochage des vésicules à la membrane plasmique. t-SNARE, v-SNARE: protéines SNARE (SNAP [soluble NSF attachment protein] receptors). Les v-SNARE sont ancrées à la membrane vésiculaire (VAMP2, synaptotagmine), tandis que les t-SNARE sont ancrées à la membrane plasmique cible (syntaxine, SNAP-25); SERCA3: calcium-ATPase de type 3 qui assure le pompage des ions Ca2+ par le réticulum endoplasmique de la cellule β; AC: adénylate-cyclases; Gs, Gq: protéines G hétérotrimériques assurant le couplage des récepteurs à sept domaines transmembranaires avec, respectivement, l’adénylate-cyclase (cas du récepteur du GLP-1) ou la phospholipase C (PLC) (cas du récepteur M3 de l’acétylcholine); Glut 2: transporteur de glucose.
Le modèle de rat GK, reproduisant spontanément les caractéristiques principales du diabète de type 2, apparaît comme un outil essentiel pour l’étude du déterminisme de la maladie et en particulier de sa composante génétique. L’homogénéité génétique de cette lignée GK, le contrôle strict de son environnement (notamment nutritionnel) et la possibilité de pratiquer des croisements informatifs avec d’autres lignées facilitent les recherches visant à identifier les facteurs génétiques et d’environnement responsables de la maladie. Cet article établit le bilan des données actuellement disponibles relatives au déterminisme de la pathologie des cellules β de la lignée GK.
Les responsables de l’atteinte β -cellulaire chez le rat GK: le jeu de l’inné et de l’acquis
L’environnement métabolique diabétique (gluco-lipotoxicité)
Chez le rat GK, l’hyperglycémie basale (assortie d’une hypertriglycéridémie modeste) apparaît seulement après le sevrage [5]. Or, c’est précisément le moment où l’anomalie sécrétoire (perte de la réponse au glucose) des cellules β GK devient détectable. Il existe donc une corrélation chronologique entre l’acquisition de l’anomalie sécrétoire et l’exposition des cellules β à un environnement métabolique diabétique. Ces changements dans le fonctionnement β-cellulaire pourraient donc refléter une perte de l’état de différenciation des cellules β lorsqu’elles sont exposées à une hyperglycémie et/ou à une hyperlipidémie chroniques (même d’intensités modestes), un phénomène appelé gluco-lipotoxicité. Cette situation chez le jeune rat GK au moment du sevrage rappelle ce qui a été décrit chez le rat adulte non diabétique lorsqu’on pratique sur ce dernier une pancréatectomie (rat Px) à 90 %. En réponse à la pancréatectomie, on observe une poussée de régénération de cellules β par néogenèse, qui reconstitue partiellement le pool de cellules β différenciées. Ce n’est qu’une à deux semaines après l’ablation que les rats Px développent une hyperglycémie basale, en général modérée mais parfois sévère [6]. Dans ce modèle, l’expression de différents gènes (glut-2, glucokinase, m-glycérol-3-phosphate déshydrogénase, puryvate-carboxylase, VDCC, voltage dependent calcium channel, SERCA-3, sarco/endoplasmic reticulum Ca2+-ATPase-3) importants pour le contrôle de l’exocytose de l’insuline induite par le glucose s’effondre progressivement avec l’apparition de l’hyperglycémie. Ces changements sont accompagnés par une diminution de l’expression de gènes codant pour des facteurs de transcription impliqués dans le développement et la différenciation des cellules β (tels que Nkx6.1, Pax6, HNF1α, HNF4α, HNF3β). En revanche, des gènes peu exprimés dans la cellule β normale (lactate-déshydrogénase-A et hexokinase-1) voient leur expression fortement accrue dans la cellule β de rat Px. Ces variations dans les niveaux d’expression des gènes sont bien induites in vivo par l’hyperglycémie elle-même, puisque le traitement des rats Px par la phlorizine (qui normalise la glycémie) pendant deux semaines ramène l’expression génique à la normale.
La programmation endocrino-métabolique
La programmation endocrino-métabolique (PEM) est définie comme un processus adaptatif se mettant en place pour répondre à une agression, souvent d’origine nutritionnelle, qui touche le mammifère à un stade vulnérable et en général précoce de son développement. La propriété tout à fait particulière de cette réponse adaptative est de donner lieu à une ou plusieurs modifications permanentes de la physiologie et du métabolisme de l’organisme, et de rester exprimée même en l’absence de l’agression/stimulus lui ayant donné naissance.
Les données obtenues sur divers modèles animaux indiquent que la PEM est un élément déterminant majeur dans la survenue d’un certain nombre de maladies adultes dégénératives [7]. L’une des illustrations les mieux documentées de ce concept de PEM est celle de l’impact sur la descendance des interventions nutritionnelles (modifiant la proportion de protéines ou d’hydrates de carbone dans la ration de la mère) appliquées, même de façon brève, pendant la période du développement. Ce type de stress nutritionnel exerce alors des effets majeurs et irréversibles sur la structure et la fonction de plusieurs organes, et en particulier du pancréas endocrine. Un exemple frappant est celui des rats nouveau-nés alimentés artificiellement avec un lait enrichi en glucides [8]: ce type d’intervention nutritionnelle limitée à la période de l’allaitement induit une adaptation du métabolisme énergétique du jeune rat qui persiste tout au long de la vie adulte, et qui crée les conditions d’apparition d’une obésité, d’une intolérance au glucose et d’anomalies de la sécrétion d’insuline. Une réduction irréversible de la réponse insulinosécrétrice au glucose est également observée chez les rats adultes primitivement exposés à la carence protéique uniquement pendant la vie foetale et/ou la période de l’allaitement [9, 10]. Le même type de PEM du pancréas endocrine a été rapporté lorsque les rats sont issus de mères rendues modérément hyperglycémiques par perfusion continue de glucose durant leur gestation [11]. Outre la fin de la vie foetale et la période d’allaitement, une PEM peut également être mis en route dès le développement embryonnaire précoce: des travaux récents viennent en effet de montrer qu’une carence en protéines (associée à une hyperglycémie très modeste) très limitée dans le temps (de E0 à E4,5 chez la rate, c’est-à-dire juste durant le développement pré-implantatoire) entraînait à très court terme un déficit de prolifération des cellules du blastocyste, source d’un retard de croissance irréversible et d’une hypertension postnatale [12]. Il convient enfin de noter que l’un des résultats, inattendus, observés dans les modèles de PEM obtenus par régime hyperglucidique pendant l’allaitement [8] ou par exposition des foetus à l’hyperglycémie maternelle [13] a été la transmission du phénotype de la 1re génération (F1) à la 2e génération (F2), sans que les foetus ou les nouveau-nés F2 n’aient subi de manipulation nutritionnelle: les rats F2 adultes manifestent ainsi un déficit de la sécrétion d’insuline. Au total, les informations fournies par les modèles de PEM qui viennent d’être cités suggèrent fortement qu’une hyperglycémie même modeste, subie pendant la vie foetale et/ou néonatale, contribue à la «programmation» du pancréas endocrine. Un tel scénario est évidemment applicable au modèle du rat GK puisque, à chaque génération, les mères GK exposent leur descendance à une hyperglycémie modérée tout au long de la gestation et de l’allaitement. Ainsi, l’hyperglycémie gestationnelle de la mère GK a pu contribuer à établir dès la F1 (en plus des gènes de susceptibilité ou même, pourquoi pas, en leur absence), puis à perpétuer au cours des générations successives (héritabilité), les anomalies du pancréas endocrine GK (Figure 4).
Figure 4
Les trois étapes possibles menant à la programmation du pancréas endocrine chez les rats exposés in utero et/ou pendant l’allaitement à des modifications nutritionnelles (hyperglycémie, carence protéique, carence énergétique).
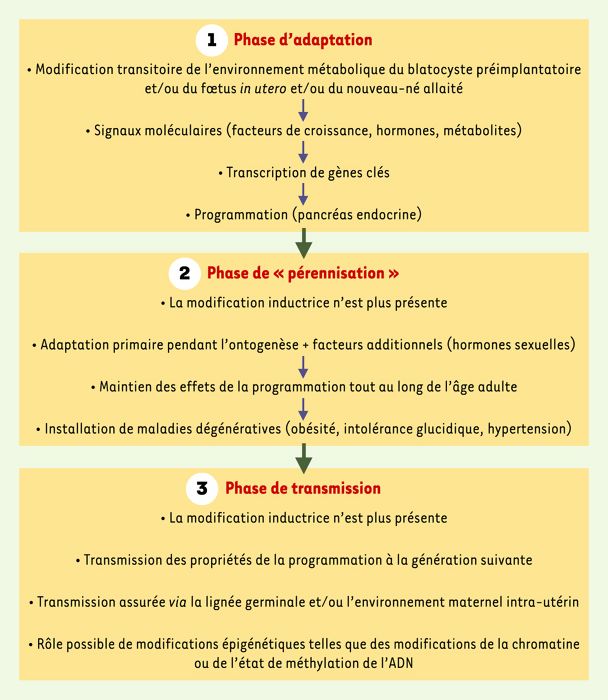
(d’après [8]).
Les mécanismes responsables de la PEM ne sont pas connus. Des mécanismes épigénétiques tels que ceux déterminant l’empreinte génomique pourraient cependant rendre compte de certains effets de la PEM [14]. Une modification épigénétique est déclenchée par des variations de facteurs d’environnement, et peut toucher au cours du développement aussi bien le lignage des cellules somatiques que les cellules germinales [15]. Par exemple, des modifications de l’état de méthylation de l’ADN ont été observées en réponse à la restriction protéique ou à la carence en folates [16]. Il est concevable et crédible de considérer que des modifications d’ordre nutritionnel appliquées au cours du développement précoce des mammifères puissent provoquer des changements spécifiques de cellule de l’état de méthylation de l’ADN nucléaire conduisant, à leur tour, à des changements spécifiques de tissu du niveau de l’expression des gènes. Enfin, et parce que les modifications spécifiques de cellule de méthylation de l’ADN sont transmises aux cellules filles lors de la réplication, les modifications initiales d’expression génique seraient ainsi immortalisées.
Les gènes de prédisposition
La stratégie utilisée pour détecter des gènes de prédisposition au diabète chez le rat GK est fondée sur l’analyse de la co-transmission, d’une génération à une autre, de certains génotypes avec le phénotype représentant la maladie. Des rats GK et des rats Brown-Norway (BN) non diabétiques ont été croisés pour obtenir une population F1 génétiquement homogène (tous les rats sont hétérozygotes). Les rats F1 ayant tous développé un diabète de sévérité modérée par rapport au rat GK, ils ont été croisés entre eux pour produire une hétérogénéité à la fois génotypique et phénotypique. Cette variabilité phénotypique en F2 est représentative du caractère polygénique du trait étudié.
L’analyse de liaison des traits phénotypiques et des génotypes par la méthode des QTL (quantitative trait locus) a permis de situer, sur 6 chromosomes différents, des gènes de contrôle de la glycémie et de l’insulinémie à jeun, de la tolérance au glucose et de la réponse insulinosécrétoire au glucose [17]. Ces différents locus ont été appelés Nidd/gk et numérotés de 1 à 6 (Nidd: non insulin dependent diabetes). Nous avons identifié sur le chromosome 2 un locus lié à l’insulinémie à jeun (Nidd/gk2), et deux locus de contrôle de la réponse insulinosécrétoire au glucose ont été localisés sur les chromosomes 4 (Nidd/gk3) et 8 (Nidd/gk5). L’établissement en cours de lignées recombinantes congéniques sélectionnées pour transposer l’un des trois locus identifiés sur le fond génétique BN devrait permettre d’identifier les composants géniques impliqués dans le déterminisme de l’atteinte β-cellulaire du rat GK.
Conclusions
L’étude menée dans le modèle GK de diabète spontané, dont le caractère polygénique est acquis, devrait être précieuse pour aider à l’identification de gènes candidats chez l’homme en orientant les efforts de recherche sur les régions humaines synténiques des locus liés au diabète GK. Elle peut aussi être utile au diabétologue pour construire de nouvelles études de liaison chez l’homme en utilisant les phénotypes associés au diabète animal qui sont spécifiquement liés aux locus identifiés chez le rat GK.
Par ailleurs, l’approche d’identification systématique des gènes dont l’expression est défectueuse, couplée à l’approche fonctionnelle permettant de tester les cascades moléculaires dans lesquelles interviennent les produits de ces gènes, devrait permettre d’éclairer un peu mieux les zones d’ombre de la physiopathologie des cellules β dans les diabètes de type 2.
Enfin, cette dernière stratégie représente une étape incontournable pour l’identification de marqueurs précoces de la maladie utilisables dans une perspective de dépistage et de médecine préventive, et pour le développement des thérapies cellulaires ciblées sur la cellule β.
Appendices
Références
- 1. Kahn SE. The importance of β-cell failure in the development and progression of type 2 diabetes. J Clin Endocrinol Metab 2001; 86; 4047-8.
- 2. Maechler P, Wolheim CB. Mitochondrial function in normal and diabetic β-cells. Nature 2001; 414; 807-12.
- 3. Neel JV. Diabetes mellitus; a «thrifty»genotype rendered detrimental by «progress»? Am J Hum Genet 1962; 14; 353-62.
- 4. Goto Y, Kakizaki M, Masaki N. Spontaneous diabetes produced by selective breeding of normal Wistar rats. Proc Jpn Acad 1975; 51; 80-5.
- 5. Portha B, Giroix MH, Serradas P, et al. β-cell function and viability in the spontaneously diabetic GK rat. Information from the GK/Par colony. Diabetes 2001; 50; 89-93.
- 6. Jonas JC, Sharma A, Hasenkamp W, et al. Chronic hyperglycemia triggers loss of pancreatic β cell differentiation in an animal model of diabetes. J Biol Chem 1999; 274; 14112-21.
- 7. Barker DJ. The fetal origins of diseases of old age. Eur J Clin Nutr 1992; 46; S3-S6.
- 8. Patel MS, Srinivasan M. metabolic programming; causes and consequences. J Biol Chem 2002; 277; 1629-32.
- 9. Blondeau B, Garofano A, Czernichow P, Bréant B. Age-dependent inability of the endocrine pancreas to adapt to pregnancy; a long-term consequence of perinatal malnutrition in the rat. Endocrinology 1999; 140; 4208-13.
- 10. Bertin E, Gangnerau MN, Bellon G, Bailbé D, Arbelot De Vacqueur A, Portha B. Development of β-cell mass in fetuses of rats deprived of protein and/or energy in last trimester of pregnancy. Am J Physiol 2002; 283; R623-R630.
- 11. Bihoreau MT, Ktorza A, Kinebanyan MF, Picon L. Impaired glucose homeostasis in adult rats from hyperglycemic mothers. Diabetes 1986; 35; 979-84.
- 12. Kwong WY, Wild AE, Roberts P, Willis AC, Fleming TP. Maternal undernutrition during the preimplantation period of rat development causes blastocyst abnormalities and programming of postnatal hypertension. Development 2000; 127; 4195-202.
- 13. Gauguier D, Bihoreau MT, Ktorza A, Berthault MF, Picon L. Inheritance of diabetes mellitus as consequence of gestational hyperglycemia in rats. Diabetes 1990; 39; 734-9.
- 14. Waterland RA, Garza C. Potential mechanisms of metabolic imprinting that lead to chronic disease. Am J Clin Nutr 1999; 69; 179-97.
- 15. Kierszenbaum AL. Genomic imprinting and epigenetic reprogramming; unearthing the garden of forking paths. Mol Reprod Dev 2002; 63; 269-72.
- 16. Jacob RA. Folate, DNA methylation, and gene expression; factors of nature and nurture. Am J Clin Nutr 2000; 72; 903-4.
- 17. Gauguier D, Froguel P, Parent V, et al. Chromosomal mapping of genetic loci associated with non-insulin dependent diabetes in the GK rat. Nat Genet 1996; 12; 38-43.
List of figures
Figure 1
Le rat GK (désigné par les initiales du nom des chercheurs Goto et Kakizaki ayant établi cette lignée) est un modèle de diabète spontané de type 2 sans obésité associée.
Cette lignée de rat a été obtenue par des croisements sélectifs de rats normaux, de souche Wistar, sélectionnés sur la base d’une légère intolérance au glucose. Cette dernière a été quantifiée par la somme (Σ glycémies) des valeurs de glycémies lors d’un test de surcharge glucosée administrée par voie orale (OGTT). La répétition de cette procédure sur de multiples générations (plus de 30) a permis d’obtenir un état diabétique stable [4]. Les animaux de la sous-lignée GK/Par sont les descendants de rats GK appartenant à la F35 de la lignée japonaise originelle [5].
Figure 2
Chronologie d’apparition et d’évolution du diabète spontané du rat GK.
DNID: diabète non insulinodépendant.
Figure 3
Voies de signalisation altérées dans la cellule β du rat GK diabétique.
Les anomalies spécifiques de la cellule β GK sont indiquées par le symbole «étoile». Le glucose est phosphorylé (G6P) par la glucokinase (GK) et converti en pyruvate (Pyr) au cours de la glycolyse. Le pyruvate entre préférentiellement dans les mitochondries et alimente la voie des acides tricarboxyliques (cycle de Krebs). Il en résulte un transfert accru d’équivalents réduits à la chaîne respiratoire, ce qui crée les conditions d’une hyperpolarisation de la membrane interne mitochondriale et d’une synthèse accrue d’ATP. C’est la fermeture ultérieure des canaux K+ dépendant de l’ATP (KIR 6.2//SUR 1) qui dépolarise la membrane plasmique. Cela va provoquer l’ouverture de canaux membranaires Ca2+ de type L dépendant du potentiel (VDCC), à l’origine de l’élévation brusque de la concentration cytosolique Ca2+ qui, in fine, activera l’exocytose des vésicules d’insuline (d’après [5]). Il existe également toute une série de signaux cytosoliques additionnels qui participent au couplage métabolisme intracellulaire-sécrétion: ce sont des composés phosphorylés, nucléotidiques ou non (ATP, GTP, AMPc, IP3), ou des métabolites (malonyl-CoA/acyl-CoA, glutamate). Ces signaux contribuent globalement à amplifier le mécanisme déclenchant décrit ci-dessus et interviennent dans des étapes plutôt distales de l’exocytose: ils modulent le fonctionnement du cytosquelette et l’accrochage des vésicules à la membrane plasmique. t-SNARE, v-SNARE: protéines SNARE (SNAP [soluble NSF attachment protein] receptors). Les v-SNARE sont ancrées à la membrane vésiculaire (VAMP2, synaptotagmine), tandis que les t-SNARE sont ancrées à la membrane plasmique cible (syntaxine, SNAP-25); SERCA3: calcium-ATPase de type 3 qui assure le pompage des ions Ca2+ par le réticulum endoplasmique de la cellule β; AC: adénylate-cyclases; Gs, Gq: protéines G hétérotrimériques assurant le couplage des récepteurs à sept domaines transmembranaires avec, respectivement, l’adénylate-cyclase (cas du récepteur du GLP-1) ou la phospholipase C (PLC) (cas du récepteur M3 de l’acétylcholine); Glut 2: transporteur de glucose.
Figure 4
Les trois étapes possibles menant à la programmation du pancréas endocrine chez les rats exposés in utero et/ou pendant l’allaitement à des modifications nutritionnelles (hyperglycémie, carence protéique, carence énergétique).
(d’après [8]).