Article body
Le terme retard mental regroupe différentes affections caractérisées selon l’OMS (Organisation mondiale de la santé) par un quotient intellectuel inférieur à 70 (QI < 70) [1]. Le retard mental est le handicap majeur le plus fréquent puisque sa prévalence est de 1 % si on ne considère que le retard mental sévère (QI < 50), et s’élève à 2-3 % si l’on inclut le retard mental léger (QI entre 50 et 70) [2, 3]. Malgré les récents progrès de la génétique médicale, dans 40 % des cas, la cause exacte du retard mental demeure cependant encore totalement inexpliquée.
On distingue classiquement deux groupes de maladies: (1) les retards mentaux syndromiques, qui associent un retard mental et des anomalies neurologiques, morphologiques, viscérales ou biochimiques; et (2) les retards mentaux non syndromiques, définis par un retard mental isolé sans autres anomalies cliniques. Les gènes impliqués dans des retards mentaux syndromiques sont très divers et participent à de multiples processus cellulaires de sorte quest souvent difficile de comprendre le lien exact entre le défaut moléculaire et la déficience cognitive observée. À l’inverse, les gènes responsables de retards mentaux non syndromiques participent directement aux processus liés aux fonctions cognitives: mémorisation, apprentissage, comportement, etc.
Le nombre des gènes responsables de retards mentaux est estimé à environ 300 et seuls 90 d’entre eux ont été identifiés. En effet, l’extrême hétérogénéité clinique et génétique de ces maladies rend difficile l’utilisation des techniques classiques d’analyse de liaison génétique, en particulier dans le cas des retards mentaux non syndromiques. En conséquence, seuls onze gènes responsables de retards mentaux non syndromiques liés à l’X ont été identifiés [4-6], mais aucun gène associé à un retard mental isolé récessif autosomique ((→) m/s 2000, n° 3, p. 363). La gravité et la fréquence de ces affections font du démembrement des gènes responsables de ces maladies un des enjeux majeurs de la génétique médicale.
Afin d’identifier certains des gènes autosomiques, nous avons choisi d’utiliser une méthode de localisation utilisant l’homozygotie par filiation. Cette stratégie, décrite en 1987 par E.S. Lander et D. Botstein, consiste à étudier des familles consanguines « multiplex » [7]. Dans de telles familles, tous les sujets atteints sont homozygotes par descendance non seulement pour la mutation morbide mais aussi pour les marqueurs avoisinants. D’autres régions sont également retrouvées homozygotes, mais sont différentes d’un individu atteint à l’autre. L’étude, par cette stratégie, d’une famille consanguine comprenant quatre enfants atteints de retard mental sévère et quatre enfants sains nous a permis de localiser le premier gène de retard mental autosomique récessif sur le bras long du chromosome 4. Parmi les nombreux gènes portés par cette partie du chromosome, nous nous sommes plus particulièrement intéressés au gène PRSS12.
Ce gène, très fortement exprimé dans le cerveau, code pour la neurotrypsine, un membre de la famille des protéases à sérine [8]. Des études menées chez la souris suggéraient que cette protéine puisse être impliquée dans les processus de maturation synaptique et de plasticité neuronale [9]. Nous avons donc déterminé la structure du gène humain et analysé, chez un des patients, l’ensemble de la séquence des treize exons codants. Ce travail nous a permis d’identifier une délétion de quatre paires de base. Cette mutation, qui conduit à un décalage du cadre de lecture et à l’apparition d’un codon d’arrêt prématuré de la traduction, co-ségrège dans cette famille, selon le mode récessif autosomique, avec le phénotype de retard mental [10]. Enfin, l’étude d’autres familles consanguines nous a permis d’identifier la même mutation dans une seconde fratrie.
Pour mieux comprendre la fonction de cette protéine, nous avons analysé son expression au cours du développement humain et dans le cerveau adulte. Nous avons montré que ce gène est fortement exprimé au cours du développement du système nerveux, en particulier dans le cortex, l’hippocampe et le noyau moteur du tronc cérébral, c’est-à-dire des structures liées à l’apprentissage et à la mémoire. Dans le cerveau adulte, la neurotrypsine s’accumule dans des vésicules synaptiques localisées à proximité de la membrane pré-synaptique des neurones corticaux (Figure 1A). Ces données nous permettent de proposer l’hypothèse selon laquelle la neurotrypsine, qui est une protéine sécrétée, se comporterait comme un neurotransmetteur en activant, par protéolyse, un récepteur spécifique ancré dans la membrane post-synaptique (Figure 1B). L’inactivation du gène de la neurotrypsine conduirait donc à un défaut de transmission synaptique par absence de la protéine.
Figure 1
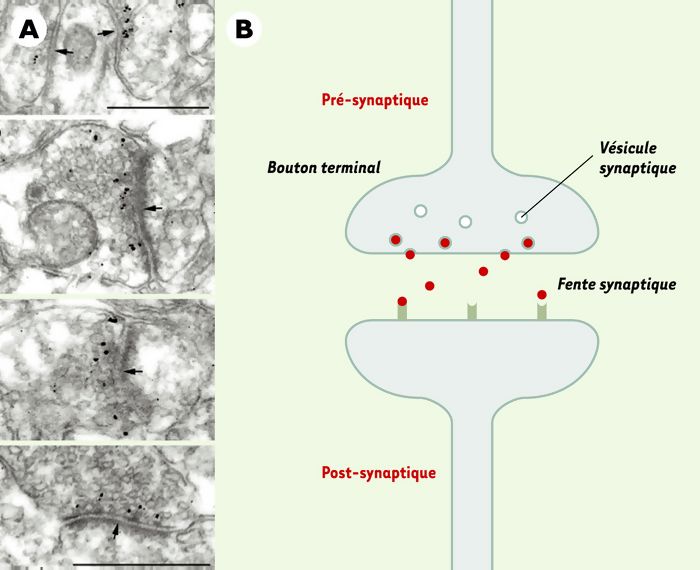
A. Localisation de la neurotrypsine dans les synapses corticales par immunohistochimie et observation en microscopie électronique. Les flèches indiquent la membrane pré-synaptique (barre = 0,5 μm). B. Représentation schématique d’une synapse montrant le mécanisme d’action possible de la neurotrypsine. La protéine (rouge) est stockée dans des vésicules synaptiques localisées à proximité de la membrane synaptique. À la suite d’une dépolarisation, la protéine est libérée dans la fente synaptique où elle pourrait reconnaître, et activer par clivage, un récepteur transmembranaire localisé dans la membrane post-synaptique. Cette activation assurerait alors la transmission du message.
Outre le fait qu’il s’agit du premier gène de retard mental non syndromique autosomique caractérisé, ces résultats ouvrent de nombreuses perspectives dans le domaine de la neurobiologie. Il s’agit en effet de la première démonstration qu’une anomalie de la protéolyse synaptique puisse conduire à un déficit intellectuel. Des travaux antérieurs, menés chez le rat et chez la souris, avaient déjà montré le rôle des protéases à sérine et de leurs inhibiteurs dans le développement et le fonctionnement du système nerveux central. Ainsi, des anomalies des synapses et des neurones sont observées chez des souris dont le gène codant pour la neuropsine, une autre protéase à sérine, est inactivé [11]. De même, plusieurs groupes ont démontré l’effet de la surexpression, ou au contraire, de l’inactivation du gène codant pour la nexine (un inhibiteur de protéase à sérine) sur la plasticité synaptique de l’hippocampe [12]. Nos résultats soulignent cependant le rôle crucial de la protéolyse synaptique dans les fonctions cérébrales humaines. Ils ouvrent ainsi la voie à d’autres travaux visant à caractériser la cascade de gènes impliqués dans ce mécanisme synaptique et leur rôle éventuel dans d’autres retards mentaux.
D’un point de vue médical, la caractérisation de la mutation à l’origine de cette maladie est une avancée majeure pour les familles concernées. Le diagnostic de ces affections permet en effet d’éviter d’autres investigations coûteuses et parfois pénibles à supporter pour l’enfant et les parents. Il fournit les éléments essentiels pour proposer un conseil génétique. Enfin, il permet de donner des éléments de réponse quant à l’évolution du handicap et aux possibilités de prises en charge thérapeutiques et/ou éducatives.
Appendices
Remerciements
Ce travail n’aurait pu être mené à bien sans l’étroite interaction de notre unité de recherche avec le département de Génétique Médicale de l’Hôpital Necker-Enfants Malades et le soutien financier de la Fondation de France et de la Fondation France Telecom.
Références
- 1. Luckasson R, Coulter DL, Polloway EA. Mental retardation: definition, classification and systems of supports. Washington DC: American Association on Mental Retardation, 1992.
- 2. Flint J, Wilkie AOM. The genetics of mental retardation. Br Med Bull 1996; 52: 453-64.
- 3. Raynham H Gibbons R, Flint J, Higgs D. The genetic basis for mental retardation. Q J Med 1996; 89: 169-75.
- 4. Chelly J, Mandel JL. Monogenic causes of X-linked mental retardation. Nat Rev Genet 2001; 2: 669-80.
- 5. Stromme P, Mangelsdorf ME, Shaw MA, et al. Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat Genet 2002; 30: 441-5.
- 6. Bienvenu T, Poirier K, Friocourt G, et al.ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Hum Mol Genet 2002; 11: 981-91
- 7. Lander ES, Botstein D. Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science 1987; 236: 1567-70.
- 8. Gschwend TP, Krueger SR, Kozlov SV, Wolfer DP, Sonderegger P. Neurotrypsin, a novel multidomain serine protease expressed in the nervous system. Mol Cell Neurosci 1997; 9: 207-19.
- 9. Wolfer DP, Lang R, Cinelli P, Madani R, Sonderegger P. Multiple roles of neurotrypsin in tissue morphogenesis and nervous system development suggested by the mRNA expression pattern. Mol Cell Neurosci 2001; 18: 407-33.
- 10. Molinari F, Rio M, Meskenaite V, et al. Truncating neurotrypsin mutation in autosomal recessive nonsyndromic mental retardation. Science 2002; 298: 1779-81.
- 11. Hirata A, Yoshida S, Inoue N, et al. Abnormalities of synapses and neurons in the hippocampus of neuropsin-deficient mice. Cell Neurosci 2000; 17: 600-10.
- 12. Luthi A, Van der Putten H, Botteri FM, et al. Endogenous serine protease inhibitor modulates epileptic activity and hippocampal long-term. J Neurosci 1997; 17: 4688-99.
List of figures
Figure 1
A. Localisation de la neurotrypsine dans les synapses corticales par immunohistochimie et observation en microscopie électronique. Les flèches indiquent la membrane pré-synaptique (barre = 0,5 μm). B. Représentation schématique d’une synapse montrant le mécanisme d’action possible de la neurotrypsine. La protéine (rouge) est stockée dans des vésicules synaptiques localisées à proximité de la membrane synaptique. À la suite d’une dépolarisation, la protéine est libérée dans la fente synaptique où elle pourrait reconnaître, et activer par clivage, un récepteur transmembranaire localisé dans la membrane post-synaptique. Cette activation assurerait alors la transmission du message.