Abstracts
Résumé
L’entrée en mitose des cellules eucaryotes est accompagnée de profondes modifications de leur organisation. L’étude de systèmes cellulaires modèles, ovocytes ou levures, a permis d’établir que l’activation des protéine kinases cycline B-cdc2 est nécessaire et apparemment suffisante pour le déclenchement de la mitose. Toutefois, les mécanismes responsables de l’activation de ces kinases dans les cellules somatiques des eucaryotes supérieurs restent très mal connus. Dans cet article, nous analysons les mécanismes possibles et montrons qu’un processus d’évolution vers la mitose, d’abord réversible, puis irréversible, est mis en route après achèvement de la duplication des chromosomes et des centrosomes. L’activation des kinases cycline B-cdc2 marque la limite entre ces deux phases. Avant cette limite, des mécanismes de surveillance, activés par divers signaux résultant d’anomalies structurales de la cellule, permettent de retarder ou de supprimer l’entrée en mitose.
Summary
Dramatic changes of cell organisation occur at onset of mitosis. Genetic analysis of fission yeast and physiological studies of vertebrate and invertebrate oocytes showed that activation of cyclin B-cdc2 kinase triggers mitosis. Nevertheless, upstream mechanisms responsible for this activation remain largely unknown in somatic cells of higher eukaryotes. This review discusses possible pathways and mechanisms involved in triggering onset of mitosis in such cells, including inhibitory checkpoint mechanisms that detect defects in structural organisation of the cell.
Article body
Le cycle cellulaire des eucaryotes présente une alternance entre deux phases, S (synthèse) et M (mitose). La phase S est une phase de duplication: celle des chromosomes, dont l’ADN se réplique par un processus semi-conservatif, permettant la production de deux chromatides qui restent associées jusqu’à la transition métaphase-anaphase de la mitose; celle également des centrosomes, qui se dupliquent par un processus mal compris, mais radicalement différent. Au début de la phase M, les centrosomes se séparent après avoir subi un processus de maturation qui augmente leur potentiel de nucléation des microtubules. Après la rupture de l’enveloppe nucléaire (chez les eucaryotes supérieurs), les paires de chromatides condensées s’associent à un appareil mitotique largement constitué de microtubules, dont la bipolarité résulte de la séparation des centrosomes. Cet appareil assure, au cours de l’anaphase, la ségrégation des deux chromatides d’une même paire dans l’un ou l’autre de deux territoires distincts, qui se séparent au cours du clivage de la cellule en deux cellules-filles.
La phase S doit être terminée avant la ségrégation des chromatides, faute de quoi une perte d’information génétique serait observée dans les cellules-filles: la phase M ne commence donc jamais avant l’achèvement de la phase S. De fait, il s’agit là de la seule contrainte absolue exercée sur le cycle cellulaire, et dans les cellules embryonnaires de nombreux métazoaires, la phase M suit immédiatement la phase S. Dans la plupart des cellules, toutefois, on observe un délai (G2) entre la phase S et la phase M, au cours duquel se produit une série de réactions intermédiaires sur lesquelles peuvent s’exercer des freins.
Ces freins peuvent être activés en réponse à des signaux extracellulaires, émis par exemple par les autres cellules du même tissu. Ils peuvent l’être également en réponse à des événements délétères se produisant dans la cellule en division (par exemple, certains dommages causés à l’ADN). Le processus qui commence par la détection de tels événements et se termine par la production d’un frein efficace constitue un mécanisme de surveillance du cycle (checkpoint). Les réactions intermédiaires sur lesquelles s’exercent ces freins n’appartiennent pas au checkpoint et sont en fait des événements normaux de la phase G2 du cycle cellulaire: ces réactions ont lieu même dans les cellules embryonnaires où la durée de la phase G2 est réduite à l’extrême [1].
Dans cet article, nous examinons les mécanismes qui, à l’issue de la phase G2, conduisent au déclenchement de la mitose chez les eucaryotes supérieurs.
Contribution de quelques système modèles à la compréhension du déclenchement de la phase M du cycle cellulaire
Ovocytes
Une des contributions majeures de l’ovocyte à l’analyse du déclenchement de la phase M a été la démonstration d’une intense vague de phosphorylations augmentant la teneur nette des cellules en nombreuses phosphoprotéines, due à l’augmentation d’activité de plusieurs protéine kinases, parmi lesquelles les kinases cycline B-cdc2 (ou cycline B-CDK1: kinase dépendante des cyclines dont la sous-unité régulatrice est la cycline B). Les cyclines mitotiques (A et B) tirent ce nom de leur dégradation périodique au cours du cycle cellulaire, leur accumulation suivant une loi en dents de scie dont la période est celle du cycle cellulaire. Chez la plupart des espèces, en particulier les quatre espèces les plus utilisées pour les recherches sur le cycle cellulaire (xénope, étoile de mer, palourde et souris), les ovocytes contiennent, dès la phase G2, les complexes cycline B-cdc2 en quantité suffisante, et sont parfaitement capables d’entrer en phase M en l’absence de nouvelle synthèse de cyclines B [2, 3]. Pendant la phase G2, les complexes cycline B-cdc2 sont maintenus sous forme inactive par phosphorylation de cdc2 sur les acides aminés thréonine 14 et tyrosine 15 (Figure 1), la thréonine/tyrosine kinase responsable de ces phosphorylations inhibitrices étant Myt1 (kinase associée aux membranes du réticulum endoplasmique et phosphorylant les résidus inhibiteurs Y15 et T14), membre de la famille Wee1 (Wee1 n’est pas exprimé dans l’ovocyte en G2). L’activation des complexes cycline B-cdc2 requiert l’activité d’une phosphatase de la famille Cdc25 (Cdc25C), capable de déphosphoryler T14 et Y15. Elle a lieu même en l’absence de noyau dans les ovocytes, et ne requiert donc pas de translocation nucléaire, l’accumulation nucléaire des complexes cycline B-cdc2 n’ayant lieu qu’après leur activation dans le cytoplasme [4-6]. Cette accumulation est nécessaire à la rupture de l’enveloppe nucléaire qui supprime, à la fin de la prophase, la compartimentation nucléocytoplasmique.
Figure 1
Déclenchement de la transition G2/M au cours du premier cycle méiotique des ovocytes.
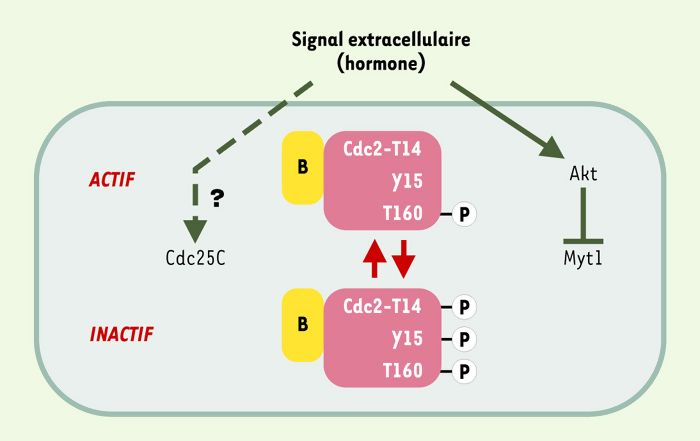
Chez la plupart des espèces, cette transition requiert la conversion de cycline B-Cdc2 d’une forme inactive, phosphorylée sur T14 et Y15, en une forme active déphosphorylée sur ces résidus. Elle est déclenchée par un signal extérieur, souvent hormonal, qui contrôle l’équilibre entre phosphorylations inhibitrices et déphosphorylations activatrices. Il a très récemment été montré chez l’étoile de mer (et pas encore étendu à d’autres espèces) que ce signal utilise la kinase Akt comme relais pour inactiver Myt1 [49]. La phosphorylation de T160, catalysée par CAK (CDK-activating kinase) stabilise les complexes cycline-CDK et est nécéssaire à leur activité [50].
La fusion d’une cellule somatique en phase G2 avec une cellule en mitose provoque l’entrée prématurée en mitose de la cellule interphasique [7]. Les ovocytes ont permis l’analyse expérimentale de ce caractère dominant de l’état mitotique en introduisant la notion de MPF (maturation-promoting factor, ou M-phase-promoting factor) et le test fonctionnel associé: lorsque du cytoplasme est prélevé d’un ovocyte sorti de G2 et micro-injecté dans un ovocyte arrêté en G2, il induit la transition G2/M dans l’ovocyte receveur [4], même en l’absence de synthèse protéique. Différents rapports ont conduit à identifier MPF à cycline B-cdc2 ((→) m/s 2001, n° 11, p. 1226) (pour revue, voir [8, 9]). La micro-injection de cette kinase purifiée sous sa forme active provoque en effet l’activation, dans l’ovocyte de xénope receveur, de la kinase inactive qu’il contient en G2. Une analyse minutieuse montre pourtant que l’activité cycline B-cdc2, bien que nécessaire, n’est souvent pas suffisante à provoquer la transition G2/M. Chez l’étoile de mer, par exemple, la kinase cycline B-cdc2 micro-injectée s’inactive en quelques secondes sous l’action de Myt1 dans le cytoplasme de l’ovocyte receveur, même si l’activité initiale de la kinase excède largement celle d’un ovocyte en phase M [10]: puisqu’elle ne s’inactive pas dans le receveur lorsqu’elle provient, non purifiée, du cytoplasme d’un ovocyte donneur en phase M, celui-ci doit contenir un facteur distinct qui s’oppose à l’inactivation par Myt1, ou qui inactive cette kinase inhibitrice. Toujours chez l’étoile de mer, l’amplification du MPF (activation du pool de cycline B-cdc2 contenu dans le receveur) requiert la présence d’un facteur d’origine nucléaire (non identifié) dans le cytoplasme du donneur, bien que la kinase cycline B-cdc2 puisse s’activer par simple stimulation hormonale dans un ovocyte G2 énucléé.
Levures
Un autre système modèle - la levure Schizosaccharomyces pombe - a été utilisé pour analyser la transition G2/M dans les conditions de la mitose.
Dans la forme sauvage de cette levure, la croissance est essentiellement associée à la phase G2, et la transition G2/M a lieu lorsque la cellule en croissance a atteint une taille standard. À température restrictive, les mutants thermosensibles cdc25 continuent à croître sans se diviser. Au contraire, les mutants wee1 se divisent à une taille inférieure à la taille standard: c’est donc par l’intermédiaire de l’équilibre Wee1/Cdc25, déterminant lui-même l’activité de la kinase Cdc2, que la croissance contrôle la transition G2/M [9] (il n’existe pas d’équivalent de Myt1 chez les levures). Au cours de la phase G2, aucune variation d’activité spécifique de la kinase inhibitrice Wee1 ou de la phosphatase activatrice Cdc25 n’a été décrite. Puisque le pool de cycline B ne cesse de croître pendant la phase G2, on pourrait penser que la transition G2/M est déclenchée lorsque la taille de ce pool atteint une valeur seuil qui dépend de la croissance. Cependant, il n’en est rien car la surexpression de la cycline B ne déclenche pas la transition G2/M de façon anticipée, c’est-à-dire à taille réduite (phénotype wee) [11].
Chez les organismes unicellulaires, la taille est une caractéristique de l’espèce, au moins dans les conditions de milieu permettant la croissance. Dans le cas de la levure S. pombe, où l’essentiel de la croissance cellulaire a lieu pendant la phase G2, on pense que les kinases Cdr1/Nim1 et Cdr2, capables de phosphoryler et d’inhiber Wee1, sont peut-être impliquées dans le contrôle nutritionnel et la relation à la croissance de l’activité MPF [12]. Néanmoins, malgré une analyse génétique approfondie du couplage entre croissance cellulaire et activation de MPF, on ignore à peu près tout du déterminisme de ce couplage. Comment la cellule mesure-t-elle sa taille? Comment utilise-t-elle cette information pour produire un signal conduisant à l’activation de MPF? Les réponses à ces questions ne sont pas connues.
Limites des systèmes modèles
Dans le cycle cellulaire « standard » des cellules somatiques, on n’observe pas d’arrêt en G2, et tandis que la cellule progresse dans cette phase, elle continue de croître. Par ailleurs, la transition G2/M a lieu et la cellule entre en mitose « spontanément », apparemment sans qu’un signal extracellulaire de déclenchement soit nécessaire à cette transition (à de rares exceptions près). De plus, l’activation des complexes cycline B-CDK1 au moment de la transition G2/M est toujours précédée par celle des complexes cycline A-CDK2.
Les ovocytes présentent des caractéristiques bien différentes: après une phase S précoce souvent limitée au développement embryonnaire de l’individu, ces cellules s’arrêtent en G2 pendant une période prolongée qui peut dans certaines espèces atteindre plusieurs dizaines d’années, en augmentant de taille pendant une partie de la période d’arrêt: même au terme de sa croissance, l’ovocyte reste verrouillé en G2. Par ailleurs, il ne subit la transition G2/M qu’après réception d’un signal extracellulaire spécifique, souvent hormonal, dont l’effet primaire est d’inactiver Myt1 (Figure 1). Enfin, les cyclines de type A ne sont souvent exprimées qu’après l’entrée des ovocytes en phase M, et non en réponse directe au signal de sortie de G2 [13]: les kinases dépendantes des cyclines A ne sont donc pas impliquées dans la transition G2/M.
Cette transition a lieu dans des conditions plus proches des cellules somatiques des eucaryotes supérieurs lorsqu’on considère la levure Schizosaccharomyces pombe. Dans ce cas, en effet, la croissance est étroitement couplée à la progression du cycle cellulaire, et la transition G2/M n’est pas déclenchée par un facteur extracellulaire. Cependant, les levures n’expriment qu’une seule forme de Cdk et la transition G2/M ne fait intervenir qu’une seule cycline, de type B. Quant à la levure Saccharomyces cerevisiae, son cycle ne comporte pas de phase G2 caractérisée, ce qui l’éloigne encore plus des eucaryotes supérieurs.
Même si les mécanismes responsables de l’entrée en phase M dans les ovocytes et la levure S. pombe présentent un caractère universel, il faut se garder d’extrapoler sans nuance au cycle cellulaire somatique des organismes complexes les conclusions déduites des études réalisées avec ces systèmes simplifiés. Chez les organismes multicellulaires, en effet, non seulement la croissance cellulaire est très étroitement couplée à la prolifération cellulaire au niveau de tous les organes, mais la taille des différents organes est fixée dans d’étroites limites au sein de l’ensemble de l’organisme. Les relations de dépendance entre croissance cellulaire et prolifération cellulaire sont donc encore plus complexes, et à peine commencent-elles à être étudiées dans un espace à trois dimensions [14, 15].
La transition G2/M dans les cellules somatiques des eucaryotes supérieurs
Activation de cycline B-CDK1 par déphosphorylation de CDK1
Comme les levures ou les ovocytes, les cellules somatiques des eucaryotes supérieurs sont irréversiblement induites à entrer en mitose lorsque les kinases cycline B-cdc2 sont activées. Dans la plupart des cellules, ces kinases sont de deux types selon la nature de la sous-unité cycline, B1 ou B2, qui diffèrent par leur extrémité amino-terminale. La cycline B1 joue un rôle essentiel, et des souris dont le gène de la cycline B1 a été invalidé meurent in utero. Au contraire, les souris dont le gène de la cycline B2 a été invalidé sont viables, quoique de taille et de fécondité un peu réduites [16]. La cycline B1 fait la navette entre le noyau et le cytoplasme pendant l’interphase. À la fin de la prophase, elle s’accumule brusquement dans le noyau, puis se lie à l’appareil mitotique. Au contraire, la cycline B2 est liée à l’appareil de Golgi, aussi bien en interphase qu’en mitose [17].
La co-expression de cycline B1 et d’un mutant de CDK1 ne pouvant être inactivé par phosphorylation (Ala14Phe15 au lieu de Thr14Tyr15) dans des cellules en culture en phase G1 du cycle cellulaire provoque prématurément de nombreux événements normalement associés à la phase M [17]: la lamina nucléaire qui double intérieurement l’enveloppe nucléaire est rapidement solubilisée (mais l’enveloppe nucléaire ne disparaît pas), les microtubules se réorganisent pour former un gros aster et les chromosomes se condensent prématurément tandis que la cellule s’arrondit. Ces changements sont associés à la translocation du complexe cycline B1-CDK1 dans le noyau. Au contraire, la co-expression de cycline B2-CDK1AlaPhe ne provoque aucun changement détectable des chromosomes, de la lamina, ni des microtubules, et les cellules restent aplaties et adhérentes au support. En revanche, cycline B2-CDK1AlaPhe et cycline B1-CDK1AlaPhe sont également efficaces pour désassembler l’appareil de Golgi et le disperser sous forme de petites vésicules cytoplasmiques.
Ces résultats impliquent que l’activation des complexes cycline B1-CDK1 par déphosphorylation des résidus inhibiteurs de CDK1 est suffisante pour déclencher la plupart des événements cytologiques de la mitose. Ils indiquent que lamina nucléaire, microtubules, chromosomes et appareil de Golgi sont des cibles directes ou indirectes des complexes cycline B-CDK. L’analyse fonctionnelle de constructions chimériques montre que les différences observées entre la surexpression de la cycline B1 et celle de la cycline B2 sont dues à des localisations intracellulaires différentes et non à des différences de spécificité intrinsèques des complexes kinasiques. De nombreux substrats de complexes cycline B-CDK ont été identifiés, incluant lamines, condensines, certains moteurs moléculaires (comme la kinésine Eg5 nécessaire à l’assemblage du fuseau mitotique) ou encore des protéines impliquées dans le contrôle de la transition G2/M telles que Cdc25C.
Mécanisme d’activation des kinases cycline B-CDK1
Au cours de la phase G2, les cellules synthétisent les cyclines B sans les dégrader et accumulent donc des complexes cycline B-CDK1. Ces complexes sont inactivés au fur et à mesure de leur production par phosphorylation inhibitrice de CDK1, catalysée par les kinases inhibitrices Wee1 et Myt 1. Par ailleurs, la micro-injection de la kinase cycline A-CDK2 dans des cellules HeLa en G2 provoque de façon anticipée l’entrée en mitose [18], tandis que la micro-injection d’anticorps dirigés contre la cycline A, ou celle d’un fragment de l’inhibiteur CIP1 (qui en phase G2 titre spécifiquement les complexes cycline A-CDK) bloque l’entrée en mitose [18, 19]. Ces résultats indiquent que l’activité de la kinase cycline A-CDK2 (peut-être aussi cycline A-CDK1) est un facteur limitant pour l’entrée en mitose. Dès que la kinase cycline B1-CDK1 est activée, sous l’effet d’une phosphatase Cdc25, l’activité cycline A-CDK2 n’est plus nécessaire. On peut donc penser que la kinase cycline A-CDK2 est requise pour l’activation des complexes cycline B1-CDK1, eux-mêmes responsables de la plupart des événements de la mitose. Cependant, la kinase cycline A-CDK2 est déjà active en phase S, alors que l’activation des kinases cycline B-CDK1 n’a lieu qu’à la fin de la phase G2. La kinase Polo qui phosphoryle et active Cdc25C [20] est également requise pour l’activation de cycline B-CDK1 dans les cellules somatiques. La déplétion de cette kinase supprime l’activation de cycline B1/2-CDK1 dans des extraits cellulaires, sans affecter l’activité des complexes cycline A-CDK [21].
Bien que la rupture de l’enveloppe nucléaire, précédée de très peu par la phosphorylation des lamines et la dépolymérisation de la lamina nucléaire, soit souvent prise comme repère pour fixer le début de la mitose, cet événement, qui requiert la translocation et l’accumulation nucléaire de la kinase cycline B1-CDK1 sous sa forme active, n’est pas le premier événement cytologique détectable de la mitose [22]. Il est précédé par des modifications structurales et fonctionnelles des centrosomes (maturation) dans le compartiment cytoplasmique et des chromosomes dans le compartiment nucléaire.
L’un des premiers événements de la maturation des centrosomes semble être l’augmentation du recrutement de la γ-tubuline, nécessaire à l’assemblage des asters de microtubules autour des centrosomes [23]. Or, la micro-injection d’anticorps inactivant la kinase Polo, ou l’expression de mutants dominants négatifs, supprime le recrutement de la γ-tubuline par les centrosomes [24]. La stimulation du recrutement de la γ-tubuline précédant l’activation de cycline B-CDK1, il doit nécessairement en aller de même pour l’activation de la kinase Polo. Celle-ci semble dépendre de phosphorylations multiples, mais son mécanisme est mal connu. Quant à la condensation des chromosomes, elle atteint son degré maximal après recrutement des condensines, lui-même corrélé chez les eucaryotes supérieurs au relargage massif de protéines spécialisées dans le maintien de la cohésion entre les chromatides, les cohésines [25]. Cette condensation est déclenchée au cours de la phase G2, en partie sous l’effet de la kinase Aurora B, phosphorylant l’histone H3 [26]: elle précède donc au moins partiellement l’activation de la kinase cycline B-cdc2. L’activité de la kinase Polo semble nécessaire à la libération des cohésines [27].
Il est actuellement difficile de dresser un scénario reliant les kinases cycline A-CDK2 et Polo à l’activation de cycline B-CDK1 (Figure 2A). La kinase cycline A-CDK2 ne semble pas capable d’activer directement Cdc25 [29]. La kinase Polo phosphoryle et active Cdc25C à la transition G2/M et l’activation de Ccd25C est synchrone de l’activation de cycline B-CDK1. Cependant, les souris dont le gène de Cdc25C a été invalidé n’ont pas de phénotype [28], impliquant que Cdc25A, ou plus vraisemblablement B, activés avant Cdc25C, sont capables de déphosphoryler efficacement CDK1 [29]). Mais rien ne permet d’exclure que les cibles primaires (majeures) de cycline A-CDK2 ou/et de la kinase Polo puissent être les kinases inhibitrices Wee1/Myt 1…
Figure 2
Déclenchement de la transition G2/M dans les cellules somatiques d’eucaryotes supérieurs.
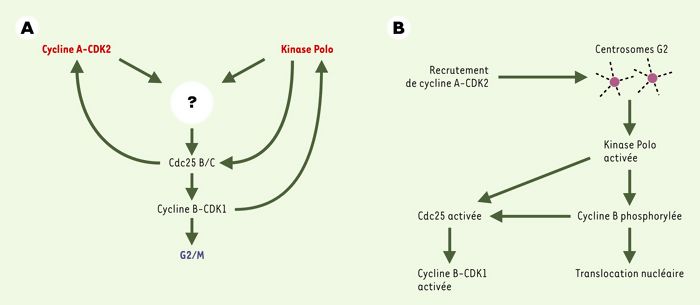
A. Les kinases cycline A-CDK2 et Polo contrôlent, par un processus intégré non élucidé, l’activation des phosphatases Cdc25 B/C requises pour la déphosphorylation des résidus inhibiteurs de CDK1 et l’activation du MPF (maturation-promoting factor). B. Un modèle (très) spéculatif d’activation de cycline B-CDK1 au niveau des centrosomes: le recrutement transitoire de la kinase cycline A-CDK2 par les centrosomes est supposé nécessaire à l’activation de cycline B-CDK1 déclenchée au niveau des centrosomes. La kinase Polo y jouerait un rôle charnière en phosphorylant et en activant Cdc25C et en phosphorylant la cycline B: cet événement faciliterait l’interaction de Cdc25B et C avec le substrat cycline B-CDK1 et - par masquage du signal d’exclusion nucléaire - l’accumulation nucléaire du complexe activé.
Sans exception connue, l’activation de cycline B1-CDK1 in vivo est associée à la phosphorylation de la cycline sur plusieurs sites conservés au cours de l’évolution [30]. Les anticorps dirigés contre les formes phosphorylées de la cycline reconnaissent donc spécifiquement la forme active des complexes cycline B1-CDK1. À l’aide de tels anticorps, il a pu être montré récemment que l’activation de cycline B-CDK1 commence au niveau des centrosomes. Au cours de la phase G2, la cycline A n’est détectée qu’au niveau du noyau. De façon intéressante, une association transitoire de la cycline A aux centrosomes a été observée en prophase [19, 31]. Cela laisse entrevoir la possibilité que le recrutement de la kinase cycline A-CDK1 par les centrosomes, au moment où ils sont également associés à la kinase Polo, joue un rôle important dans l’activation des complexes cycline B-CDK1 au niveau de cet organite essentiel pour la progression du cycle cellulaire. Les centrosomes constitueraient alors la structure au sein de laquelle est réalisée l’intégration des processus qui contribuent au déclenchement de la mitose. Contrairement à celle des cyclines B, la synthèse/accumulation de la cycline A est un facteur limitant pour la transition G2/M [11, 18]. Une analyse détaillée du processus conduisant au recrutement transitoire de la cycline A par les centrosomes pourrait ouvrir de nouvelles perspectives dans notre analyse de la transition G2/M. Un modèle possible, très spéculatif, est présenté dans la Figure 2B.
Dans les exemples qui précèdent, un processus d’évolution vers la mitose, d’abord réversible, puis irréversible, est mis en route après achèvement de la duplication des chromosomes et des centrosomes. Dans certains cas, cependant, ce processus n’a pas lieu, et les cellules recommencent à dupliquer les chromosomes, devenant alors polyploïdes [32] ((→) m/s 2002, n° 12, p. 1219). Puisque la croissance continue et qu’elles ne se divisent pas, ces cellules augmentent de taille. Le processus d’endoréplication est souvent associé à une simplification du cycle cellulaire: par exemple, certaines cellules suppriment la mitose en n’exprimant pas CDK1 ou ses activateurs, cycline B, cycline A ou CDC25C. De façon intéressante, certaines cellules qui « endorépliquent » ont perdu les centrosomes [33], donnant encore plus de poids à l’hypothèse selon laquelle ces structures jouent un rôle déterminant dans le déclenchement de la mitose.
Cdc25 B/C et Polo, cibles des checkpoints G2/M
Des traitements ou des événements provoquant l’arrêt de la progression des fourches de réplication et des cassures de l’ADN [34], des anomalies de « décaténation » des chromatides-soeurs après réplication [35] ((→) m/s 2002, n° 3, p. 282), la dépolymérisation des microtubules [36] et peut-être certaines modifications structurales de la matrice nucléaire [37] peuvent avoir pour conséquence l’arrêt du cycle cellulaire en phase G2, dû à l’activation d’un checkpoint G2/M. Lorsqu’elles ont activé la kinase cycline B-CDK1, les cellules somatiques deviennent insensibles au checkpoint [18] et sont irréversiblement induites à entrer en mitose (même si celle-ci s’avère défectueuse). Avant cette activation, en revanche, certains traitements sont capables, non seulement d’arrêter la progression du cycle, mais de faire régresser les cellules, provoquant par exemple le relargage de γ-tubuline par les centrosomes [36] et la décondensation des chromosomes [37]. A priori, l’activation des checkpoints G2/M pourrait provoquer l’arrêt en G2 en supprimant, par exemple, l’expression de la cycline A ou en augmentant l’activité des kinases inhibitrices Wee1/Myt1. Dans l’état actuel des connaissances, cependant, les cibles des checkpoints G2/M semblent plutôt être les phosphatases Cdc25 B/C et la kinase Polo, dont l’activation est supprimée (Figure 3) (des résultats récents suggèrent également un rôle pour l’inhibiteur CIP1) ((→) m/s 2001, n° 3, p. 353).
Figure 3
Cdc25 B/C et Polo, cibles des checkpoints G2/M.

Les phosphatases Cdc25B et C et la kinase Polo sont les principales cibles connues des checkpoints G2/M activés par les UV, les drogues dépolymérisant les microtubules, les agents tels qu’hydroxyurée ou rayonnements ionisants bloquant la réplication ou provoquant des cassures de l’ADN. On ignore actuellement quelle peut être la cible du checkpoint de « décaténation », bien que sa suppression par mutation du signal d’exclusion nucléaire de la cycline B1 suggère un rôle possible de la kinase Polo.
La suppression de l’activation des phosphatases Cdc25 B/C fait intervenir la consolidation de leur association avec divers membres de la famille 14-3-3. Il est possible que l’interaction de Cdc25 avec 14-3-3 masque certains résidus requis pour l’interaction avec la cycline B [38], empêchant l’interaction de cycline B-CDK1 avec le site catalytique de Cdc25. La liaison de 14-3-3 s’oppose également à la translocation nucléaire des phosphatases [39]. Cette liaison requiert la phosphorylation de la sérine 216 (Cdc25C humain) ou de la sérine 309 (Cdc25 B humain). Au cours du cycle cellulaire normal, ces résidus sont phosphorylés jusqu’à la fin de la phase G2, la kinase responsable de ces phosphorylations étant cTak1 [40]. À la transition G2/M, ces résidus sont déphosphorylés et 14-3-3 libéré. Lorsque les checkpoints G2/M liés à l’interruption de la réplication de l’ADN, les cassures des brins d’ADN, ou les défauts de « décaténation » des chromatides-soeurs sont activés, d’autres kinases viennent renforcer c-Tak1, rendant impossible la déphosphorylation des résidus inhibiteurs de Cdc25 B/C. Ces kinases sont Chk1 et Chk2 (encore appelées Cds1). De façon remarquable, les kinases Chk1 et Chk2 semblent incapables de phosphoryler Cdc25 B/C lorsque ces phosphatases ont pris leur conformation mitotique, après activation de MPF. Ces kinases ont peut-être une activité basale constitutive, mais ne subissent une activation notable qu’après phosphorylation par les kinases ATM et ATR [34, 40], qui elles-mêmes transduisent les stimulus initiaux.
Il est possible d’inhiber les kinases ATM et ATR par la caféine, et de renforcer l’inhibition des checkpoints dépendant de ces kinases par un nouvel inhibiteur de Chk1, UCN-01. Même dans ces conditions, les radiations ionisantes et les rayons ultratviolet (UV) provoquent un arrêt en G2 [40]. Ces traitements agissent en activant la kinase de stress p38 [41]. En effet, son inhibition supprime le checkpoint, et sa micro-injection l’active en l’absence d’UV. Or, la kinase p38 phosphoryle Cdc25B sur deux sites responsables après phosphorylation de la liaison de 14-3-3, cette phosphorylation n’ayant pas lieu après irradiation si la kinase p38 est inhibée. Il semble très probable que l’inhibition de Cdc25B a pour conséquence l’inactivation de cycline A-CDK2, car l’expression ectopique du mutant non phosphorylable CDK2-Ala14Phe15 associé à cycline A lève le bloc dépendant de G2 de la kinase p38. Nous avons proposé plus haut que l’activation de la kinase cycline B-CDK1 puisse être déclenchée au niveau des centrosomes par une régulation concertée faisant intervenir la kinase cycline A-CDK2 et la kinase Polo. Il est possible que le checkpoint G2/prophase contrôle la fonction de déclenchement de la mitose au niveau des centrosomes. L’arrêt en G2 provoqué par la kinase de stress p38 pourrait représenter une « mesure de précaution », permettant d’arrêter par anticipation la progression du cycle cellulaire, même si les rayonnements potentiellement dangereux n’ont pas provoqué de dégâts nucléaires: dans ce cas, l’arrêt n’est que transitoire. Au contraire, il se prolonge si la voie ATM/ATR est activée.
La kinase Polo semble être une autre cible des checkpoints G2/M. D’une part, la micro-injection d’anticorps dirigés contre cette kinase arrête en phase G2 des cellules non immortalisées, sans empêcher l’entrée en mitose des cellules transformées [24]. D’autre part, un composant essentiel du checkpoint G2/M, Chfr, a été récemment identifié à une ligase qui ubiquitinyle la kinase Polo [42] et provoque sa protéolyse (dans les extraits d’oeufs de xénope). Le gène Chfr est très fréquemment muté, ou non exprimé, dans les cellules tumorales dont le checkpoint G2/M est défectueux [43], l’expression ectopique de Chfr restaurant le checkpoint. La compartimentation cellulaire de cycline B1-CDK1 semble également impliquée dans les checkpoints G2/M. En effet, l’expression d’une cycline B1 contenant une mutation du signal d’export nucléaire supprime, dans les cellules HeLa, l’arrêt en G2 provoqué par les défauts de « décaténation » [44]. Or, dans le cycle cellulaire normal, l’inactivation du signal d’export nucléaire de la cycline B1 requiert sa phosphorylation par la kinase Polo [39]. Le checkpoint G2/M de « décaténation », indépendant de Chk1 et de cdc25 B/C, pourrait donc agir en supprimant l’activité de la kinase Polo.
Parallèlement à Chfr, une autre ubiquitine-ligase [45], BRCA1 (sous forme d’un complexe avec BARD1) joue un rôle essentiel dans les checkpoints activés par les radiations ionisantes ou les défauts de « décaténation ». En phase G2, cette ubiquitine-ligase est normalement concentrée au niveau des focus nucléaires [45] et des centrosomes [46]. Quand les cellules sont soumises à un stress génotoxique, elle se redistribue des focus nucléaires vers différents sites cellulaires, où elle exerce vraisemblablement sa fonction d’ubiquitinylation. Les protéines ciblées par l’ubiquitine-ligase BRCA1, probablement en vue de leur dégradation par les protéasomes, n’ont pas fait l’objet d’un inventaire exhaustif. Parmi elles figure la grande sous-unité de l’ARN polymérase 2 [47], dont le recrutement par l’ubiquitine-ligase fait intervenir le facteur Cst50, impliqué dans le contrôle de la polyadénylation des ARN messagers.
Conclusions
Dans cet article, nous avons tenté de cerner les mécanismes responsables du déclenchement de la mitose. Une première constatation est qu’il n’est pas aisé de définir de façon précise le début de la mitose. Certains événements tels que la condensation des chromosomes commencent bien avant la rupture de l’enveloppe nucléaire et la mise en place de l’appareil mitotique. Par ailleurs, la condensation des chromosomes est réversible jusqu’à un point de non-retour, au-delà duquel la cellule ne peut revenir en interphase et entrera nécessairement en mitose, même si elle reçoit des signaux d’activation du checkpoint G2/prophase. Ce point de non-retour est atteint lorsque la kinase MPF (cycline B-cdc2) est activée: il est donc logique de prendre cet événement comme « temps zéro » de la mitose. L’étude de la transition G2/M devient alors celle du processus d’activation du MPF, la condensation des chromosomes s’effectuant en partie en phase G2. S’il semble probable que l’activation du MPF commence au niveau des centrosomes, il reste difficile de préciser si la maturation de ceux-ci commence en phase G2 ou si elle coïncide avec la transition G2/M.
Pendant la phase G2, le potentiel d’inactivation du MPF domine, malgré la synthèse de cycline B qui augmente progressivement le niveau du complexe cycline B-cdc2. L’activité de la kinase cycline A-CDK2 est un facteur limitant de la transition G2/M. Il est toutefois difficile de rendre compte du caractère brutal de l’entrée en mitose par l’augmentation progressive d’activité de la kinase cycline A-CDK2 au cours de la phase G2. Il est possible que la transition G2/M soit déclenchée par le recrutement au niveau des centrosomes de la kinase cycline A-CDK2, cela n’ayant lieu qu’après titration de ses sites de liaison nucléaires. L’activation de la kinase Polo, essentielle pour la maturation des centrosomes, l’est également pour cette transition. On ignore actuellement encore si cette activation requiert elle-même le recrutement de la kinase cycline A-CDK2 par les centrosomes et, d’une manière plus générale, comment le recrutement programmé des opérateurs par le centrosome conduit à l’activation initiale du MPF au niveau de cet organite, dont la micro-injection dans des ovocytes d’étoile de mer arrêtés en G2 au terme de la maturation méiotique suffit à induire la transition G2/M [48].
Des mécanismes de surveillance (checkpoints G2/M) permettent d’arrêter de façon plus ou moins durable la progression du cycle cellulaire avant l’activation de cycline B-CDK1. Utilisant des voies de signalisation cytoplasmiques ou nucléaires, ces checkpoints semblent exercer leur effet, au moins en partie, en empêchant l’activation des phosphatases Cdc25 B/C et celle de la kinase Polo, qui contrôle non seulement l’activation de Cdc25C, mais également la phosphorylation de la cycline B1 et la translocation nucléaire de la kinase cycline B1-CDK1.
Appendices
Références
- 1. Kim SH, Li C, Maller JL. A maternal form of the phosphatase Cdc25 A regulates early embryonic cell cycles in Xenopus laevis. Dev Biol 1999; 212: 381-91.
- 2. Abrieu A, Dorée M, Fisher D. The interplay between cycle B-cdc2 kinase (MPF) and MAP kinase during maturation of oocytes. J Cell Sci 2001; 114: 257-67.
- 3. Hochegger H, Klotzbucher A, Kirk J, et al. New-type cyclin synthesis is required between meiosis I and meiosis II during Xenopus oocyte maturation. Development 2001; 128: 3795-807.
- 4. Masui Y, Markert CL. Cytoplasmic control of nuclear behavior during meiotic maturation of frog oocytes. J Exp Zool 1971; 177: 129-45.
- 5. Picard A, Harricane MC, Labbé JC, Dorée M. Germinal vesicle components are not required for the cell cycle oscillator of the early starfish embryo. Dev Biol 1988; 128: 121-8.
- 6. Okata K, Hiranaga S, Okano T, Tachibana K, Kishimoto T. Relocation and distinct subcellular localization of p34cdc2-cyclin B complex at meiosis reinitiation in starfish oocytes. EMBO J 1992; 11: 1763-72.
- 7. Johnson RT, Rao PN. Mammalian cell fusion: induction of premature chromosome condensation in interphase nuclei. Nature 1970; 226: 717-22.
- 8. Nurse P. Universal control mechanisms regulating onset of M-phase. Nature 1990; 344: 503-8.
- 9. Dorée M, Hunt T. From Cdc2 to Cdk1: when did the cell cycle kinase join its cyclin partner? J Cell Sci 2002; 115: 2461-4.
- 10. Picard A, Labbé JC, Barakat H, Cavadore JC, Dorée M. Okadaic acid mimics a nuclear component required for cyclin B-cdc2 kinase microinjection to drive starfish oocytes into M-phase. J Cell Biol 1991; 115: 337-44.
- 11. Booher R, Beach D. Involvement of cdc13+ in mitotic control in Schizosaccharomyces pombe: possible interaction of the gene product with microtubules. EMBO J 1988; 7: 2321-7.
- 12. Belenguer P, Pelloquin L, Baldin V, Oustrin ML, Ducommun B. The fission yeast Nim1/Cdr1 kinase: a link between nutritional state and cell cycle control. Prog Cell Cycle Res 1995; 1: 207-14.
- 13. Okano-Uchida T, Sekiai T, Lee K, Okumura E, Tachibana K, Kishimoto T. In vivo regulation of cyclin A-cdc2 and cyclin B-cdc2 through meiotic and early cleavage cycles in starfish. Dev Biol 1998; 197: 39-53.
- 14. Su TT, O’Farrell PH. Cell proliferation does not equal growth. Curr Biol 1998; 8: R687-9.
- 15. Potter CJ, Xu T. Mechanisms of size control. Curr Opin Genet Dev 2001; 11: 279-86.
- 16. Brandeis M, Rosewell I, Carrington M, et al. Cyclin B2-null mice develop normally and are fertile whereas cyclin B1-null mice die in utero.Proc Natl Acad Sci USA 1998; 95: 434-9.
- 17. Draviam VM, Orrechia S, Lowe M, Pardi R, Pines J. The localization of human cyclins B1 and B2 determines CDK1 substrate specificity and neither enzyme requires MEK to disassemble the Golgi apparatus. J Cell Biol 2001; 152: 945-58.
- 18. Furuno J, den Elzen N, Pines J. Human cyclin A is required for mitosis until mid prophase. J Cell Biol 1999; 147: 295-306.
- 19. Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin A is required at two points in the human cell cycle. EMBO J 1992; 11: 961-71.
- 20. Kumagai A, Dunphy WG. Purification and molecular coloning of Plx1, a Cdc25-regulatory kinase fom Xenopus egg extracts. Science 1996; 273: 1377-80.
- 21. Abrieu A, Brassac T, Galas S, Fisher D, Labbé JC, Dorée M. The Polo-like kinase Plx1 is a component of the MPF amplification loop at the G2/M-phase transition of the cell cycle in Xenopus eggs. J Cell Sci 1998; 111: 1751-7.
- 22. Pines J, Rieder CL. Re-staging mitosis: a contemporary view of mitotic progression. Nat Cell Biol 2001; 3: E3-6.
- 23. Do Carmo Avides M, Tavares A, Glover DM. Polo kinase and Asp are needed to promote the mitotic organizing activity of centrosomes. Nat Cell Biol 2001; 3: 421-4.
- 24. Lane HA, Nigg EA. Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J Cell Biol 1996; 135: 1701-13.
- 25. Lozada A, Hirano T. Shaping the metaphase chromosome: coordination of cohesion and condensation. Bioessays 2001; 23: 924-35.
- 26. Giet R, Glover DM. Drosophila aurora B kinase is required for histone H3 phosphorylation and condensin recruitment during chromosome condensation and to organize the central spindle during cytokinesis. J Cell Biol 2001; 152: 669-82.
- 27. Alexandre G, Uhlmann F, Mechtler K, Poupart MA, Nasmyth K. Phosphorylation of the cohesin subunit Scc1 by Polo/Cdc25 kinase regulates sister chromatid separation in yeast. Cell 2001; 105: 459-72.
- 28. Chen MS, Hurov J, White LS, Woodford-Thomas T, Piwnica-Worms H. Absence of apparent phenotype in mice lacking Cdc25c protein phosphatase. Mol Cell Biol 2001; 21: 3853-61.
- 29. Lammer C, Wagerer S, Saffrich R, Mertens S, Ansorge W, Hoffmann I. The Cdc25 phosphatase is essential for the G2/M phase transition in human cells. J Cell Sci 1998; 111: 2445-53.
- 30. Peter M, Le Peuch C, Labbé JC, Meyer A, Donoghue DJ, Dorée M. Initial activation of cyclin-B1-cdc2 kinase requires phosphorylation of cyclin B1. EMBO Rep 2002; 3: 551-6.
- 31. Bailly E, Pines J, Hunter T, Bornens M. Cytoplasmic accumulation of cyclin B1 in human cells: association with a detergent-resistant compartment and with the centrosome. J Cell Sci 1992; 101: 529-45.
- 32. Edgar BA, Orr-Weaver TL. Endoreplication cell cycles: more for less. Cell 2001; 105: 297-306.
- 33. Mahowald AP, Caulton JH, Edwards MK, Floyd AD. Loss of centrioles and polyploïdization in follicle cells of Drosophila melanogaster. Exp Cell Res 1979; 118: 404-10.
- 34. Rhind N, Russell P. Chk1 and Cds1: linchpins of the DNA damage and replication checkpoint pathways. J Cell Sci 2000; 113: 3889-96.
- 35. Deming PB, Cistulli CA, Zhao H, et al. The human decatenation checkpoint. Proc Natl Acad Sci USA 2001; 98: 12044-9.
- 36. Rieder CL, Cole R. Microtubule disassemby delays the G2-M transition in vertebrates. Curr Biol 2000; 10: 1067-70.
- 37. Rieder CL, Cole RW. Entry into mitosis in vertebrate somatic cells is guarded by a chromosome damage checkpoint that reverses the cell cycle when triggered during early but not late prophase. J Cell Biol 1998; 142: 1013-22.
- 38. Morris MC, Heitz A, Mery J, Heitz F, Divita GB. An essential phosphorylation-site domain of human CDC25C interacts with both 14-3-3 and cylins. J Biol Chem 2000; 37: 28849-57.
- 39. Takizawa CG, Morgan DO. Control of mitosis by changes in the subcellular location of cyclin B1-Cdk1 and Cdc25C. Curr Opin Cell Biol 2000; 12: 658-65.
- 40. Bulavin DV, Amundson SA, Fornace Jr AJ. p38 and Chk1 kinases: different conductors for the G2/M checkpoint symphony. Curr Opin Genet Dev 2002; 12: 92-7.
- 41. Bulavin DV, Higashimoto Y, Popoff IJ, et al. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinae. Nature 2001; 411: 102-7.
- 42. Kang D, Chen J, Wong J, Fang G. The checkpoint potein Chfr is a ligase that ubiquitinates Plk1 and inhibits Cdc2 at the G2 to M transition. J Cell Biol 2002; 156: 249-59.
- 43. Scolnick DM, Halazonetis TD. Chfr defines a mitotic stress checkpoint that delays entry into metaphase. Nature 2000; 406: 430-5.
- 44. Toyoshima F, Moriguchi T, Wada A, Fukuda M, Nishida E. Nuclear export of cyclin B1 and its possible role in the DNA damage-induced G2 checkpoint. EMBO J 1998; 17: 2728-35.
- 45. Baer R, Ludwig T. The BRCA1/BARD1 heterodimer, a tumor suppressor complex with ubiquitin E3 ligase activity. Curr Opin Genet Dev 2002; 12: 86-96.
- 46. Hsu LC, White RL. BRCA1 is associated with the centrosome during mitosis. Proc Natl Adac Sci USA 1998; 95: 12983-8.
- 47. Chiba N, Parvin JD. Redistribution of BRCA1 among four different protein complexes following replication blockage. J Biol Chem 2001; 14: 38549-54.
- 48. Picard A, Karsenti E, Dabauvalle, MC, Dorée M. Release of mature starfish oocytes from interphase arrest by microinjection of human centrosomes. Nature 1987; 327: 170-2.
- 49. Okumura E, Fukubara T, Yoshida H, et al. Akt inhibits Myt in the signalling pathway that leads to meiotic G2/M phase transition. Nat Cell Biol 2002; 4: 111-6.
- 50. Fesquet D, Morin N, Dorée M, Devault A. Is Cdk7/cyclin H/MAT1 the genuine cdk activating kinase in cycling Xenopus oocytes? Oncogene 1997; 15: 1303-7.
List of figures
Figure 1
Déclenchement de la transition G2/M au cours du premier cycle méiotique des ovocytes.
Chez la plupart des espèces, cette transition requiert la conversion de cycline B-Cdc2 d’une forme inactive, phosphorylée sur T14 et Y15, en une forme active déphosphorylée sur ces résidus. Elle est déclenchée par un signal extérieur, souvent hormonal, qui contrôle l’équilibre entre phosphorylations inhibitrices et déphosphorylations activatrices. Il a très récemment été montré chez l’étoile de mer (et pas encore étendu à d’autres espèces) que ce signal utilise la kinase Akt comme relais pour inactiver Myt1 [49]. La phosphorylation de T160, catalysée par CAK (CDK-activating kinase) stabilise les complexes cycline-CDK et est nécéssaire à leur activité [50].
Figure 2
Déclenchement de la transition G2/M dans les cellules somatiques d’eucaryotes supérieurs.
A. Les kinases cycline A-CDK2 et Polo contrôlent, par un processus intégré non élucidé, l’activation des phosphatases Cdc25 B/C requises pour la déphosphorylation des résidus inhibiteurs de CDK1 et l’activation du MPF (maturation-promoting factor). B. Un modèle (très) spéculatif d’activation de cycline B-CDK1 au niveau des centrosomes: le recrutement transitoire de la kinase cycline A-CDK2 par les centrosomes est supposé nécessaire à l’activation de cycline B-CDK1 déclenchée au niveau des centrosomes. La kinase Polo y jouerait un rôle charnière en phosphorylant et en activant Cdc25C et en phosphorylant la cycline B: cet événement faciliterait l’interaction de Cdc25B et C avec le substrat cycline B-CDK1 et - par masquage du signal d’exclusion nucléaire - l’accumulation nucléaire du complexe activé.
Figure 3
Cdc25 B/C et Polo, cibles des checkpoints G2/M.
Les phosphatases Cdc25B et C et la kinase Polo sont les principales cibles connues des checkpoints G2/M activés par les UV, les drogues dépolymérisant les microtubules, les agents tels qu’hydroxyurée ou rayonnements ionisants bloquant la réplication ou provoquant des cassures de l’ADN. On ignore actuellement quelle peut être la cible du checkpoint de « décaténation », bien que sa suppression par mutation du signal d’exclusion nucléaire de la cycline B1 suggère un rôle possible de la kinase Polo.