Abstracts
Résumé
La tyrosinémie héréditaire de type 1 est la maladie la plus sévère de la voie du catabolisme de la tyrosine et est due à une perte d’activité de la fumarylacétoacétate hydrolase. Les patients souffrent principalement d’atteinte hépatique conduisant le plus souvent au cancer du foie. À partir des données récentes sur les sites d’action des métabolites toxiques, un nouveau modèle permettant d’expliquer l’étiopathogénie de la tyrosinémie héréditaire de type 1 - et, notamment, le taux élevé de cancer du foie observé - peut être proposé.
Summary
Hereditary tyrosinemia type 1 (HT1) is the most severe metabolic disease associated with tyrosine catabolism. An accumulation of toxic metabolites seems responsible for the pathology of HT1. The metabolite fumarylacetoacetate, accumulating due to a deficiency in fumarylacetoacetate hydrolase, displays apoptogenic, mutagenic, aneugenic and mitogenic activities. These effects may underlie the tumorigenic phenomenon observed in HT1. Fumarylacetoacetate in addition to causing disturbances in Ca2+ homeostasis, may induce endoplasmic reticulum stress.
Article body
La tyrosinémie héréditaire de type 1 (OMIM 276700) (TH1) est une maladie autosomique récessive [1]. Elle résulte d’un déficit de la fumarylacétoacétate hydrolase (FAH), le dernier enzyme impliqué dans la voie catabolique de la tyrosine (Figure 1). Ce déficit entraîne une accumulation de métabolites tels le fumarylacétoacétate (FAA), le maléylacétoacétate et le succinylacétone, le dosage de ce dernier servant de test diagnostique de la maladie. Il a été proposé que l’accumulation de ces métabolites soit responsable des effets toxiques hépatique et rénal et des crises neurologiques chez les patients en bas âge [1]. Deux formes cliniques de TH1 ont été décrites sur la base de la sévérité de la maladie et de l’âge du diagnostic. La forme aiguë se manifeste très tôt dans l’enfance et est caractérisée par des dommages hépatiques sévères pouvant mener à une défaillance hépatique et au décès. La forme chronique se manifeste plus tardivement et se caractérise par un dysfonctionnement tubulaire rénal, une cirrhose hépatique et, chez de nombreux patients, le développement d’hépatocarcinomes. Le traitement de la TH1 repose actuellement sur une diète restrictive en tyrosine, phénylalanine et méthionine, accompagnée d’un traitement par un inhibiteur de l’enzyme hydroxyphénylpyruvate dioxygénase, en amont de la FAH, le 2-(2-nitro-4-trifluorométhylbenzoyl)-1,3-cyclohexanedione (NTBC) [2]. Malgré ce traitement, la transplantation de foie s’avère parfois nécessaire.
Figure 1
Voie catabolique de la tyrosine.
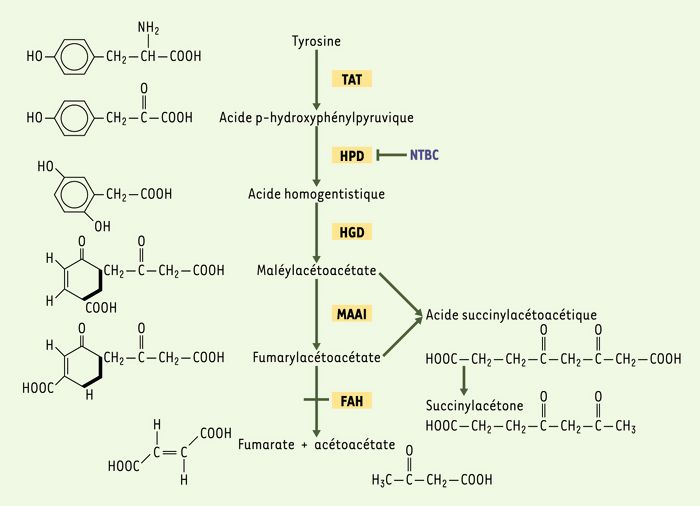
La tyrosinémie héréditaire de type 1 est due à un blocage de la voie catabolique de la tyrosine au niveau de l’enzyme fumarylacétoacétate hydrolase (FAH). HGD: acide homogentisique dioxygénase, HPD: 4-hydroxyphénylpyruvate dioxygénase, MAAI: maléylacétoacétate isomérase, NTBC: 2-(2-nitro-4-trifluorométhylbenzoyl)-1,3-cyclohexanedione, TAT: tyrosine aminotransférase.
La TH1 a été rapportée dans la plupart des régions du monde mais la région du Saguenay-Lac-St-Jean au Québec présente une incidence particulièrement élevée avec un nouveau-né atteint sur 1846 [3]. Dans cette région, la mutation d’épissage IVS12+5g → a représente 95% des mutations [1, 4]. À ce jour, 41 mutations responsables de la maladie ont été répertoriées (Figure 2) [5, 6]. Toutefois, compte tenu de l’hétérogénéité clinique observée chez des patients portant la même mutation [7], aucune corrélation n’a pu être établie entre les différentes mutations et la sévérité de la maladie. Une particularité de la TH1 est l’incidence élevée de correction du défaut génétique dans le foie des patients TH1: une expression mosaïque de la FAH, due à une réversion de mutation et à une sélection positive des cellules corrigées, a été observée chez plus de 85% des patients [7-9]. Ce même phénomène a été décrit dans un modèle murin de TH1 [10].
Figure 2
Localisation des mutations dans le gène de la fumarylacétoacétate hydrolase au cours de la tyrosinémie héréditaire de type I.
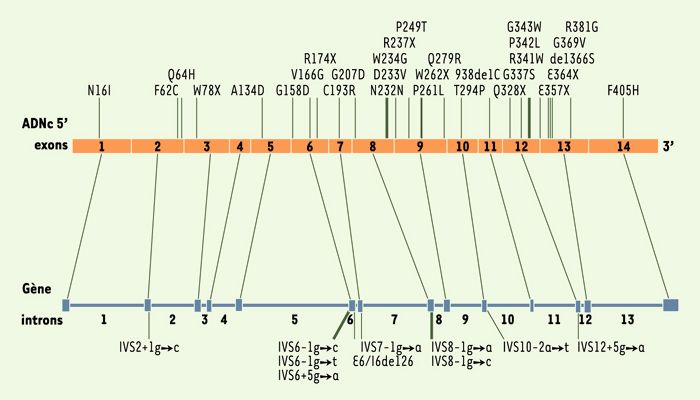
Quarante et une mutations associées à la TH1 ont été répertoriées dont 19 mutations faux-sens, 13 mutations d’épissage, 8 mutations non-sens et une mutation silencieuse.
Le fumarylacétoacétate, métabolite inducteur d’apoptose
Des études sur des cellules en culture [11] et dans un modèle animal de TH1 [12] ont suggéré que l’accumulation de FAA, liée au déficit de la FAH, induit une apoptose massive dans le foie des patients TH1 (forme aiguë). En effet, dans les cellules HeLa et V79, le FAA cause un arrêt du cycle cellulaire en G2/M suivi d’une entrée en apoptose [11]. L’apoptose induite par le FAA comprend deux événements précoces, l’activation de la cycline-B kinase et celle de la caspase-1, suivie d’un relargage mitochondrial de cytochrome c, d’une activation de la caspase-3 et finalement d’une perte du potentiel de membrane de la mitochondrie. L’inhibiteur général des caspases, z-VAD-fmk, prévient l’apoptose initiée par le FAA. Ces perturbations du cycle cellulaire sont dépendantes de la concentration de glutathion (GSH) intracellulaire et sont accentuées par une diminution du GSH. De plus, la normalisation de la concentration de GSH par le GSH-monoéthylester ou la N-acétylcystéine abolit les propriétés cytostatiques et apoptogéniques du FAA. Ni le maléylacétoacétate ni le succinylacétone ne démontrent d’activité apoptotique.
Le fumarylacétoacétate, un métabolite mutagénique et aneugénique
La fréquence élevée (37%) de cancer du foie chez les patients TH1 agés de plus de deux ans [13] ainsi que l’observation d’un pourcentage élevé de réversion de mutation dans le gène fah dans le foie [7-9], suggèrent qu’un produit de dégradation de la tyrosine puisse causer des dommages à l’ADN.
Un effet mutagénique du FAA, mais non du maléylacétoacétate ni du succinylacétone, a pu être démontré dans des cellules V79: le FAA induit trois fois plus de mutations que ce qui est observé dans les cellules témoin [14]. Les mutations sont 10 fois plus fréquentes si la concentration de GSH cellulaire a été préalablement diminuée par le L-buthionine-(S,R)-sulphoxymine [15]. Ces travaux ont donc permis d’identifier le FAA comme métabolite mutagénique et comme agent qui déplète le GSH.
L’effet mutagénique du FAA est potentialisé par une diminution du GSH cellulaire, une potentialisation qui est réversible. Il faut noter que l’activité mutagénique du FAA n’est pas liée à une formation de radicaux libres [15].
Récemment, dans des cellules humaines et de rongeurs, nous avons observé qu’une dose sublétale de FAA induit des anomalies du fuseau mitotique, des défauts de ségrégation des chromosomes et la formation de cellules multinuclées et de micronoyaux [16].
La plupart de ces micronoyaux contenait des fragments chromosomiques avec centromère (aneugenèse). Ces données expérimentales sont à rapprocher des brins chromosomiques observés dans des fibroblastes de patients TH1 [17]. De plus, il a été rapporté que l’activité de l’ADN ligase était réduite à 20 % dans les fibroblastes TH1, effet qui serait dû à l’accumulation de succinylacétone [18]. Ainsi, le processus de transformation cellulaire serait dû aux effets mutagéniques, aneugéniques et à la diminution de l’efficacité de réparation de l’ADN secondaires à l’accumulation de FAA et/ou de succinylacétone.
Le fumarylacétoacétate, un agent perturbateur de l’homéostasie du calcium intracellulaire
Associées à l’instabilité chromosomique induite par le FAA, une fragmentation du complexe de Golgi, une augmentation de la concentration intracellulaire de Ca2+ et une activation de la kinase ERK (extracellular signal-regulated kinase) ont été observées à la fois dans des modèles cellulaires [16] et dans des fibroblastes de patients TH1.
Ces observations nous permettent de proposer un nouveau modèle cellulaire d’action du FAA (Figure 3). Nous suggérons que le FAA induit un relargage de Ca2+ des réservoirs intracellulaires (appareil de Golgi et réticulum endoplasmique), ainsi qu’une augmentation de la concentration cytosolique de Ca2+ relayée par la phosphatidylinositol 3-kinase et la phospholipase C. Cette libération de Ca2+ est suffisante pour induire l’activation de la kinase ERK. L’activation de ERK dépend des radicaux thiol, de la tyrosine kinase et du complexe Ca2+/calmoduline, mais n’est pas modulée par les facteurs de croissance ni par la protéine kinase C [16].
Figure 3
Modèle d’activation de ERK (extracellular signal-regulated kinase) par le fumarylacétoacétate.
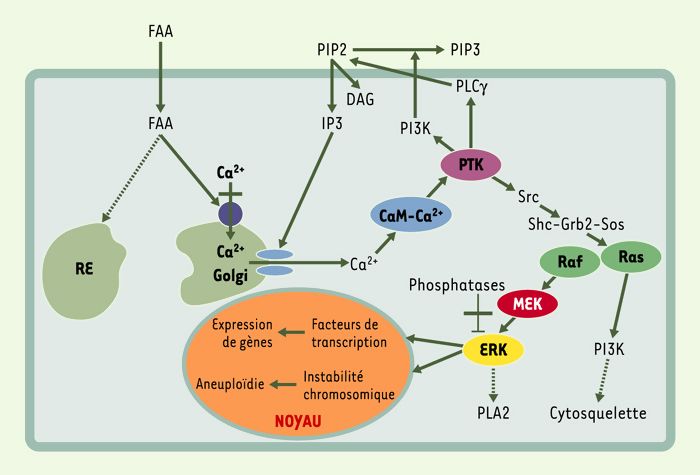
L’activation de ERK induite par le fumarylacétoacétate (FAA) suit la voie de phosphorylation classique Ras → Raf → MEK et est causée par un relargage de Ca2+ des réservoirs cellulaires tels que l’appareil de Golgi. Ceci suffit pour causer une activation de la protéine tyrosine kinase (PTK) menant à l’activation de Ras vraisemblablement via les médiateurs Src, Shc, Grb-2 et Sos. L’activation de PTK provoque également l’activation de la phosphatidylinositol 3-kinase (PI3K) et de la phospholipase γ (PLCγ) résultant en un recyclage de phosphatidylinositol 4,5 biphosphate (PIP2) et en une production d’inositol 1,4,5-trisphosphate (IP3). La production de IP3 contribue à l’augmentation de Ca2+ cytoplasmique en ciblant le récepteur de IP3 à la surface des organites qui servent de réservoirs au Ca2+. L’activation soutenue de ERK est due à un déséquilibre de la balance phosphorylation/déphosphorylation. Un relargage de Ca2+ favorise la phosphorylation de ERK alors qu’une mobilisation du Ca2+ et l’activité GSH/thiol-induite du FAA par inhibition des protéines phosphatases favorise la déphosphorylation de ERK. Une activation soutenue de ERK par le FAA pourrait contribuer à la carcinogenèse en augmentant l’expression aberrante de certains gènes, en causant une instabilité chromosomique et en provoquant des perturbations structurales (appareil de Golgi). CaM: calmoduline; RE: réticulum endoplasmique.
L’activation soutenue de ERK, causée par un déséquilibre de son état de phosphorylation/déphosphorylation, lui-même dû aux variations de concentration de Ca2+ et de GSH induites par le FAA, serait responsable des perturbations mitotiques, de l’instabilité chromosomique et d’une augmentation d’expression génique aberrante. Le cumul des effets mutagéniques, aneugéniques et mitogéniques du FAA serait à la base du processus de cancérogenèse dans le foie des patients TH1.
Le fumarylacétoacétate, inducteur de stress du réticulum endoplasmique
Des perturbations structurales de l’appareil de Golgi et du réticulum endoplasmique ont été rapportées dans des cellules du parenchyme hépatique et dans les cellules des tubules proximaux des reins de souris dont le gène codant pour la FAH a été invalidé [19]. Dans les fibroblastes des patients TH1 ainsi que dans des modèles de cellules traitées par le FAA, l’appareil de Golgi est également altéré. L’augmentation de la concentration de Ca2+ suite à un traitement au FAA provient vraisemblablement d’un relargage de Ca2+ de ces deux principaux réservoirs cellulaires. Néanmoins, nous ne pouvons exclure la possibilité que les fonctions du réticulum endoplasmique soient spécifiquement perturbées par le FAA. Deux voies de signalisation associées au stress du réticulum ont été décrites. La première, la voie de réponse aux protéines mal repliées (UPR, unfolded protein response), est activée en réponse à la présence de protéines mal repliées dans cet organite et se caractérise par l’induction de protéines chaperones du réticulum (BiP/GRP78, GRP94 ou calréticuline, par exem-ple) et du facteur de transcription CHOP/GADD153 [20, 21]. La seconde, la voie de surcharge du réticulum endoplasmique (EOR, endoplasmic reticulum overload response), est induite par une accumulation de protéines dans le réticulum menant à une libération de Ca2+ du réticulum et à la production subséquente de radicaux libres provoquant ainsi une réponse inflammatoire via l’activation du facteur de transcription NF-κB [22]. Des résultats préliminaires obtenus dans notre laboratoire semblent indiquer que le FAA cause effectivement une perturbation des fonctions du réticulum endoplasmique. L’induction de la protéine chaperone BiP/GRP78, de même que l’induction de CHOP/GADD153 par le FAA, suggèrent qu’une accumulation de FAA puisse solliciter la voie de réponse aux protéines mal repliées. Puisque le FAA ne semble pas induire la production de radicaux libres, il est peu probable que la voie de surcharge du RE soit impliquée.
Plusieurs maladies génétiques telles la fibrose kystique et certaines formes d’Alzheimer sont causées par des mutations qui mènent à l’accumulation de protéines mal repliées dans le réticulum endoplasmique, résultant en l’activation de cette voie de signalisation ou encore en l’activation de la voie de surcharge du réticulum tel que suggéré par l’expression de gènes pro-inflammatoires. Nos résultats récents suggèrent qu’un stress du RE puisse être associé à la TH1. Cet effet semblerait dû à une accumulation de FAA provoquant un «engorgement» de protéines mal repliées ou dénaturées au sein du RE.
Conclusions
Une meilleure compréhension des bases moléculaires de la TH1 est essentielle pour le développement de nouvelles thérapies car le NTBC ne supprime pas l’apparition de cancers dans un modèle murin de la TH1 [23]. Les observations selon lesquelles un rétablissement de la concentration de GSH intracellulaire atténue les effets toxiques du FAA que sont l’apoptose, la mutagenèse et la mitogenèse, suggèrent que la sévérité de la pathologie pourrait être en relation avec le degré d’accumulation de FAA et les fluctuations des concentrations de GSH intracellulaire. Ces facteurs pourraient aussi influencer le degré de réversion observé dans le foie des patients TH1. On peut ainsi envisager comme thérapie alternative ou complémentaire, d’utiliser de composés permettant d’augmenter la concentration de GSH intracellulaire dans les cellules hépatiques.
Appendices
Références
- 1. Mitchell GA, Grompe M, Lambert M, Tanguay RM. Hypertyrosinemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic and molecular bases of inherited diseases, 8e ed. New York: McGraw-Hill, 2001: 1777-805.
- 2. Lindsted S, Holme E, Lock EA, Hialmarson O, Strandvik B. Treatment of hereditary tyrosinaemia type 1 by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet 1992; 340: 813-7.
- 3. De Braekeleer M, Larochelle J. Genetic epidemiology of hereditary tyrosinemia in Quebec and in Saguenay-Lac-St-Jean. Am J Hum Genet 1990; 47: 302-7.
- 4. Poudrier J, St-Louis M, Lettre F, et al. Frequency of the IVS12+5g: a splice mutation of the fumarylacetoacetate hydrolase gene in carriers of hereditary tyrosinemia in the French Canadian population of Saguenay-Lac-St-Jean. Prenat Diagn 1996; 16: 59-64.
- 5. St-Louis M, Tanguay RM. Mutations in the fumarylacetoacetate hydrolase gene causing hereditary tyrosinemia type 1: overview. Hum Mutat 1997; 9: 291-9.
- 6. Arranz JA, Pinol F, Kozak L, et al. Splicing mutations, mainly IVS6-1(g>t), account for 70 % of fumarylacetoacetate hydrolase (FAH) gene alterations, including 7 novel mutations, in a survey of 29 tyrosinemia type I patients. Hum Mutat 2002; 20: 180-8.
- 7. Poudrier J, Lettre F, Scriver CR, Larochelle J, Tanguay RM. Different clinical forms of hereditary tyrosinemia (type 1) in patients with identical genotypes. Mol Genet Metab 1998; 64: 119-25.
- 8. Kvittingen EA, Rootwelt H, Brandtzaeg P, Bergan A, Berger R. Hereditary tyrosinemia type 1. Self-induced correction of the fumarylacetoacetase defect. J Clin Invest 1993; 91: 1816-21.
- 9. Kvittingen EA, Rootwelt H, Berger R, Brandtzaeg P. Self-induced correction of the genetic defect in tyrosinemia type 1. J Clin Invest 1994; 94: 1657-61.
- 10. Overturf K, Al-Dhalimy M, Tanguay RM, et al. Hepatocytes corrected by gene therapy are selected in vivo in a murine model of hereditary tyrosinemia type I. Nat Genet 1996; 12: 266-73.
- 11. Jorquera R, Tanguay RM. Cyclin B-dependent kinase and caspase-1 activation precedes mitochondrial dysfunction in fumarylacetoacetate-induced apoptosis. Faseb J 1999; 13: 2284-98.
- 12. Kubo S, Sun M, Miyahara M, et al. Hepatocyte injury in tyrosinemia type 1 is induced by fumarylacetoacetate and is inhibited by caspase inhibitors. Proc Natl Acad Sci USA 1998; 95: 9552-7.
- 13. Weinberg AG, Mize CE, Worthen HG. The occurrence of hepatoma in the chronic form of hereditary tyrosinemia. J Pediatr 1976; 88: 434-8.
- 14. Tanguay RM, Jorquera R, Poudrier J, St-Louis M. Tyrosine and its catabolites: From disease to cancer. Acta Biochim Pol 1996; 43: 209-16.
- 15. Jorquera R, Tanguay RM. The mutagenicity of the tyrosine metabolite, fumarylacetoacetate, is enhanced by glutathione depletion. Biochem Biophys Res Commun 1997; 232: 42-8.
- 16. Jorquera R, Tanguay RM. Fumarylacetoacetate, the metabolite accumulating in hereditary tyrosinemia, activates the ERK pathway and induces mitotic abnormalities and genomic instability. Hum Mol Genet 2001; 10: 1741-52.
- 17. Gilbert-Barness E, Barness LA, Meisner LF. Chromosomal instability in hereditary tyrosinemia type1. Pediatr Pathol 1990; 10: 243-52.
- 18. Prieto-Alamo MJ, Laval F. Deficient DNA-ligase activity in the metabolic disease tyrosinemia type I. Proc Natl Acad Sci USA 1998; 95: 12614-8.
- 19. Kelsey G, Ruppert S, Beermann F, Grund C, Tanguay RM, Schutz G. Rescue of mice homozygous for lethal albino deletions: Implications for an animal model for the human liver disease tyrosinemia type I. Genes Dev 1993; 7: 2285-97.
- 20. Spear E, Ng DT. The unfolded protein response: No longer just a special teams player. Traffic 2001; 2: 515-23.
- 21. Yoneda T, Urano F, Ron D. Transmission of proteotoxicity across cellular compartments. Genes Dev 2002; 16: 1307-13.
- 22. Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 1999; 13: 1211-33.
- 23. Al-Dhalimy M, Overturf K, Finegold M, Grompe M. Long-term therapy with NTBC and tyrosine-restricted diet in a murine model of hereditary tyrosinemia type I. Mol Genet Metab 2002; 75: 38-45.
List of figures
Figure 1
Voie catabolique de la tyrosine.
La tyrosinémie héréditaire de type 1 est due à un blocage de la voie catabolique de la tyrosine au niveau de l’enzyme fumarylacétoacétate hydrolase (FAH). HGD: acide homogentisique dioxygénase, HPD: 4-hydroxyphénylpyruvate dioxygénase, MAAI: maléylacétoacétate isomérase, NTBC: 2-(2-nitro-4-trifluorométhylbenzoyl)-1,3-cyclohexanedione, TAT: tyrosine aminotransférase.
Figure 2
Localisation des mutations dans le gène de la fumarylacétoacétate hydrolase au cours de la tyrosinémie héréditaire de type I.
Quarante et une mutations associées à la TH1 ont été répertoriées dont 19 mutations faux-sens, 13 mutations d’épissage, 8 mutations non-sens et une mutation silencieuse.
Figure 3
Modèle d’activation de ERK (extracellular signal-regulated kinase) par le fumarylacétoacétate.
L’activation de ERK induite par le fumarylacétoacétate (FAA) suit la voie de phosphorylation classique Ras → Raf → MEK et est causée par un relargage de Ca2+ des réservoirs cellulaires tels que l’appareil de Golgi. Ceci suffit pour causer une activation de la protéine tyrosine kinase (PTK) menant à l’activation de Ras vraisemblablement via les médiateurs Src, Shc, Grb-2 et Sos. L’activation de PTK provoque également l’activation de la phosphatidylinositol 3-kinase (PI3K) et de la phospholipase γ (PLCγ) résultant en un recyclage de phosphatidylinositol 4,5 biphosphate (PIP2) et en une production d’inositol 1,4,5-trisphosphate (IP3). La production de IP3 contribue à l’augmentation de Ca2+ cytoplasmique en ciblant le récepteur de IP3 à la surface des organites qui servent de réservoirs au Ca2+. L’activation soutenue de ERK est due à un déséquilibre de la balance phosphorylation/déphosphorylation. Un relargage de Ca2+ favorise la phosphorylation de ERK alors qu’une mobilisation du Ca2+ et l’activité GSH/thiol-induite du FAA par inhibition des protéines phosphatases favorise la déphosphorylation de ERK. Une activation soutenue de ERK par le FAA pourrait contribuer à la carcinogenèse en augmentant l’expression aberrante de certains gènes, en causant une instabilité chromosomique et en provoquant des perturbations structurales (appareil de Golgi). CaM: calmoduline; RE: réticulum endoplasmique.