Abstracts
Résumé
L’existence de lymphocytes T capables de reconnaître des antigènes spécifiques des tumeurs est maintenant complètement admise. Ces cellules, ainsi que les cellules natural killer (NK), infiltrent le tissu tumoral et sont en mesure d’exercer une cytotoxicité à l’égard des cellules cancéreuses. Cependant, au cours de l’histoire naturelle du développement tumoral, la réponse immunitaire se révèle inefficace. Ce constat est à l’origine de nombreux travaux de recherche dont l’objectif à terme est de concevoir de nouvelles approches thérapeutiques du cancer. L’immunothérapie a fait la preuve de son efficacité sur des modèles expérimentaux de tumeur chez l’animal. La thérapie cellulaire du cancer par cellules immunocompétentes est actuellement en évaluation clinique. Dans ces stratégies thérapeutiques, deux approches sont en évaluation : stimuler in vivo le développement d’une immunité protectrice, à l’aide de cellules présentatrices d’antigènes, ou encore apporter au patient des cellules effectrices éduquées in vitro.
Summary
The identification of tumor specific antigens has provided important advance in tumor immunology. It is now established that specific cytotoxic T lymphocytes (CTL) and natural killer cells infiltrate tumor tissues and are effector cells able to control tumor growth. However, such a natural antitumor immunity has limited effects in cancer patients. Failure of host defenses against tumor is consecutive to several mechanisms which are becoming targets to design new immunotherapeutic approaches. CTL are critical components of the immune response to human tumors and induction of strong CTL responses is the goal of most current vaccine strategies. Effectiveness of cytokine therapy, cancer vaccines and injection of cells improving cellular immunity have been established in tumor grafted murine models. Clinical trials are underway. To day, interest is particularly focused on cell therapy : injected cells are either « ready to use » effector cells (lymphocytes) or antigen presenting cells able to induce a protective immune reaction in vivo (dendritic cells). The challenge ahead lie in the careful optimization of the most promising stategies in clinical situation.
Article body
La découverte de l’immunité antitumorale est une histoire pleine de rebondissements qui a pris ses racines au XIXe siècle, lorsqu’un chirurgien du Memorial Hospital de New York, William Coley, a constaté des régressions de tumeur chez des malades qui développaient parallèlement un épisode infectieux. Un peu plus tard, d’autres équipes ont confirmé que l’injection d’extraits de bactéries à des souris pouvait entraîner la nécrose hémorragique de leur tumeur : ces substances stimulaient les défenses immunitaires de l’organisme qui, à leur tour, limitaient la prolifération cancéreuse.
Les cellules tumorales peuvent être reconnues par le système immunitaire
C’est dans ces conditions expérimentales que le TNF (tumor necrosis factor) a été découvert. D’autres arguments sont en faveur de cette immunosurveillance : par exemple, la découverte, au cours d’autopsies, de tumeurs cliniquement silencieuses, ou encore l’observation, confirmée par des données anatomopathologiques, de régressions spontanées de tumeurs. De plus, les cancers sont plus fréquents pendant la période néonatale ainsi que chez les sujets âgés, deux périodes de la vie au cours desquelles le système immunitaire serait moins efficace. Ces maladies sont également plus fréquentes chez les sujets immunodéprimés : c’est le cas, en particulier, des tumeurs associées à des virus chez des patients traités par immunosuppresseurs. Un argument encore plus probant concerne l’infiltration de certaines tumeurs par des cellules lymphoïdes (lymphocytes T et cellules natural killer, NK). Depuis longtemps, cette infiltration est considérée comme le signe d’un pronostic favorable.
La mise en évidence des antigènes tumoraux
La reconnaissance par le système immunitaire des tumeurs humaines a été démontrée par le groupe de T. Boon de l’Institut Ludwig à Bruxelles, en 1992. Les travaux expérimentaux à l’origine de cette découverte ont consisté à cultiver des cellules issues d’une biopsie de tumeur primaire, en présence des lymphocytes périphériques du même sujet (Figure 1) [1]. Au cours de cette culture mixte, les lymphocytes reconnaissant des antigènes tumoraux prolifèrent et se différencient en lymphocytes T cytotoxiques (CTL), qui peuvent être clonés. Les clones obtenus sont utilisés pour sélectionner des sous-populations de cellules tumorales dont les génomes peuvent alors être comparés. C’est ainsi que les gènes codant pour ces antigènes dits « de tumeur » ont été identifiés. Ces expériences pionnières, réalisées à partir de cellules de mélanome humain, ont permis d’identifier les gènes MAGE (melanoma antigen). La même stratégie appliquée à plusieurs types de tumeurs a permis d’identifier d’autres gènes de tumeur. Ces gènes codent pour des protéines qui peuvent être dégradées par la cellule et présentées à sa surface sous forme de peptides par l’intermédiaire des molécules d’histocompatibilité de classe I (CMH-I) (Figure 2). Un peptide est dit antigénique lorsqu’il est reconnu comme étranger par l’organisme. Des lymphocytes T spécifiques de cet antigène peuvent alors être mis en évidence. Il est immunogène lorsque cette reconnaissance se traduit par une réponse immunitaire efficace.
Figure 1
Obtention de clones CTL antitumoraux autologues.

L’expérience de clonage des lymphocytes T spécifiques des antigènes tumoraux a consisté à cultiver des cellules tumorales provenant d’une biopsie, à les irradier, puis à les mélanger à des lymphocytes circulants du même patient. Les lymphocytes dirigés contre un antigène tumoral prolifèrent et se différencient. Ils sont alors clonés et testés pour leur capacité de lyser certaines cellules tumorales issues de la culture primaire. Des sous-populations tumorales peuvent ainsi être sélectionnées et les gènes codant pour ces antigènes dits « de tumeur » sont identifiés par comparaison des génomes des différentes sous-populations (d’après [1]).
Les antigènes de tumeur chez l’homme
Plusieurs catégories d’antigènes de tumeur peuvent être distinguées, mais aucune classification n’est véritablement satisfaisante. Ils peuvent être spécifiques des cellules tumorales ou simplement associés aux tumeurs.
Les antigènes spécifiques de tumeur
Ils sont appelés antigènes « publics » lorsque leur expression est partagée par différents types histologiques de cancers. Ils peuvent être le produit d’un gène normalement présent dans le génome, mais qui ne s’exprime pas dans les cellules normales adultes. C’est le cas de la famille des gènes MAGE, BAGE (bladder antigen), GAGE (gastric antigen), RAGE (renal antigen) ou encore de l’α-foetoprotéine. Aucune expression des gènes MAGE n’est détectable dans les tissus normaux, à l’exception des testicules et du placenta, mais 75 % des mélanomes expriment au moins un des quatre gènes MAGE. On peut citer également des antigènes partagés par plusieurs types de tumeur et résultant d’un défaut de glycosylation. Dans les tumeurs mammaires, de l’ovaire ou du pancréas, une glycosylation anormale de la mucine MUC1 engendre des déterminants antigéniques nouveaux. Dans 80 % des cellules tumorales mammaires, le gène MUC1 est surexprimé ou amplifié. La protéine Muc1 peut être clivée et la fraction soluble devient l’antigène circulant Ca 15.3, un marqueur très utilisé dans le suivi des patientes atteintes de cancer du sein. Une seule copie du proto-oncogène HER-2/neu est présent dans les cellules normales, mais le gène est amplifié dans certaines cellules tumorales. La protéine HER-2/neu, à peine détectable en immunohistochimie dans les tissus adultes, est surexprimée dans environ 40 % des cancers du sein et de l’ovaire.
Une mutation ponctuelle peut également conduire à une protéine anormale donnant naissance à un peptide antigénique. Des oncogènes (ras) ou des gènes suppresseurs (p53) mutés peuvent ainsi se révéler à la fois oncogéniques et antigéniques. Par exemple, dans 50 % des cancers du côlon et 90 % des adénocarcinomes pancréatiques, les mutations de ras codent pour une protéine p21ras, connue pour son antigénicité. Il en va de même pour des mutations sur la β-caténine ou encore sur Cdk4. Des protéines cellulaires anormales (et antigéniques) peuvent également provenir de gènes viraux intégrés dans le génome de la cellule tumorale. En effet, certains virus comme le virus d’Epstein-Barr, de l’hépatite B ou encore des papillomes jouent un rôle initiateur de la cancérogenèse. De la même manière, la translocation (9 ;22) observée dans les leucémies myéloïdes chroniques qui aboutit à la fusion des gènes bcr/abl est responsable d’une protéine chimérique. La portion de cette protéine qui correspond à l’endroit de la fusion Bcr/Abl est antigénique.
Figure 2
Adressage des antigènes endogènes pour une présentation membranaire.

Les peptides issus du protéasome, ainsi que certains petits peptides cytosoliques d’origine lysosomiale, sont transportés dans la lumière du réticulum endoplasmique granuleux (REG) par un complexe de transport TAP (transporter-associated with antigen processing). Les molécules de classe I synthétisées dans le REG accueillent les peptides qui y sont transférés. Cette association est complétée par la fixation de la β2-microglobuline (β2-m, en jaune) et est chaperonnée par des protéines d’adressage (HSP). Les peptides associés aux molécules CMH-I sont ensuite véhiculés dans des vésicules recouvertes de coatomères (manteau impliqué dans la formation des vésicules adressées au réticulum andoplasmique) qui empruntent le flux vectoriel permanent. Ils se positionnent à la surface cellulaire par un mécanisme d’exocytose constitutive.
Les antigènes associés aux tumeurs
Ces antigènes peuvent être les produits de gènes de différenciation exprimés de façon limitée par les tissus sains, mais surexprimés ou amplifiés dans les tissus cancéreux correspondants. Il s’agit par exemple de protéines spécifiques des mélanocytes comme la tyrosinase, la gp100, Melan-A/MART1, gp75 ou encore de protéines ubiquitaires associées à un état prolifératif comme la télomérase. L’antigène prostatique (PSMA) ainsi que l’antigène carcino-embryonnaire (ACE) peuvent être classés dans cette catégorie. C’est le cas également des gènes RU2 et alt-M-CSF dans les cellules rénales, à ceci près que certains transcrits de RU2 (RU2A) ne sont exprimés que dans les cellules tumorales où ils sont antigéniques [2].
Des CTL spécifiques de tous les antigènes de tumeur préalablement cités ont pu être clairement mis en évidence chez des patients cancéreux, et notamment au sein des populations de lymphocytes infiltrant les tumeurs (TIL).
La réponse immunitaire antitumorale est souvent inefficace
Il est donc bien établi que les cellules tumorales peuvent être reconnues spécifiquement par des cellules effectrices du système immunitaire. Des TIL peuvent être isolés et cultivés à partir de fragments tumoraux ou de ganglions de drainage. L’évaluation fonctionnelle de ces lymphocytes permet de confirmer leur aptitude à lyser les cellules tumorales in vitro ou encore à sécréter des cytokines de type Th1 en réponse à la présentation des antigènes spécifiques. Pour que ces cellules effectrices soient efficaces dans leur fonction lytique, il faut que le contact établi entre la cellule cible qui présente l’antigène et la cellule effectrice qui possède le récepteur T spécifique de cet antigène (TCR, T cell receptor) soit stabilisé par des molécules d’adhérence complémentaires et exposées à la surface des deux types cellulaires (Figure 3). Bien qu’ils soient présents, ces effecteurs sont malgré tout souvent inefficaces in vivo : la tumeur continue de se développer et des métastases peuvent envahir les différents organes. Cela peut être dû à des défauts de reconnaissance de la cellule tumorale ou encore d’activation des cellules effectrices spécifiques. Les mécanismes moléculaires à l’origine de ces défauts fonctionnels sont nombreux. Comme toute réponse immunitaire, la réponse anti-tumorale doit être déclenchée par des cellules présentatrices d’antigènes. Les cellules tumorales sont des cellules du soi et, de ce fait, bien qu’anormales, très faiblement inductrices de réponse. Le rôle des cellules présentatrices professionnelles est alors crucial pour le développement d’une défense efficace. Nous verrons successivement les aspects théoriques d’une réponse immunitaire antitumorale idéale (phase d’induction et phase effectrice), puis les défaillances avérées de cette réponse en cas de progression tumorale in vivo.
Figure 3
Reconnaissance spécifique et activation du lymphocyte T par une cellule présentatrice d’antigène (CPA).

L’antigène présenté par les molécules du CMH (de classe II ou I) est reconnu par des lymphocytes T (CD4 ou CD8 respectivement) spécifiques de cet antigène. Les lymphocytes ne seront activés que si le signal (1) (reconnaissance spécifique du peptide) est accompagné des signaux dits de co-stimulation (2). Le second signal est délivré au lymphocyte T lors de l’interaction de récepteurs accessoires avec des ligands situés à la surface des CPA : les molécules B7 jouent ce rôle par leur liaison aux récepteurs CD28. En l’absence de ce signal 2, le lymphocyte devient tolérant à l’antigène. Différentes molécules d’adhérence stabilisent ces interactions.
Déclenchement d’une réponse immunitaire anti-tumorale « idéale »
Il a été démontré, tout au moins in vitro, que les cellules dendritiques étaient les cellules présentatrices d’antigène (CPA) les plus efficaces pour engendrer des effecteurs cytotoxiques spécifiques des cellules tumorales. Les cellules dendritiques sont des cellules hétérogènes tant sur le plan de leur origine hématopoïétique que de leur état de différenciation. Les cellules dendritiques myéloïdes immatures sont des cellules sentinelles, disséminées dans la plupart des tissus, qui présentent, comme le macrophage, une aptitude particulière aux mécanismes d’endocytose et de phagocytose. In vivo, elles internalisent les antigènes exogènes dans les tissus périphériques, puis migrent ensuite, sous l’influence de différents facteurs (chimiokines produites dans un contexte inflammatoire), jusqu’aux organes lymphoïdes secondaires. Ces antigènes peuvent provenir de lysats, de fragments ou de corps apoptotiques de cellules tumorales. Tout ceci se déroule lorsque les cellules de l’immunité innée, macrophages et cellules NK, n’ont pas été suffisamment efficaces pour éliminer toutes les cellules anormales (Figure 4). La nature des voies d’adressage intracellulaire des antigènes exogènes dans la cellule dendritique conditionne leur mode de présentation final (Figure 5). C’est dans les aires T d’un ganglion de drainage que se met en place la réponse cellulaire T. L’antigène présenté par les molécules de classe II est reconnu par des lymphocytes T CD4 spécifiques de cet antigène. Ces lymphocytes T ne seront activés que si le premier signal (reconnaissance du peptide) est accompagné des signaux dits de co-stimulation (transmis par les molécules B7, voir Figure 3). Le niveau d’expression des molécules de co-stimulation est corrélé à l’état de maturation des cellules dendritiques. Les lymphocytes T CD4 ainsi activés sécrètent des cytokines, en particulier l’IL-2 nécessaire à leur prolifération. Les cellules T CD4 favorisent également la maturation des CPA par l’engagement du CD40L (Figure 4). Dans des conditions de maturation et de contexte cytokinique (pro-inflammatoire) optimales, les cellules dendritiques deviennent capables d’activer directement des T CD8 naïfs [3]. Ce sont les seules CPA douées de cette capacité, et ceci grace à leur niveau d’expression des molécules de costimulation et de présentation. Cette activation directe permet d’engendrer une réponse cellulaire cytotoxique protectrice. Dans le cas d’une réponse immunitaire « idéale », la présence de signaux de « danger » ou de stress favorise une maturation optimale des cellules dendritiques [4]. Ces signaux peuvent être de nature microbienne (c’est ce qui se passe de manière particulièrement efficace dans le cas de la réponse immunitaire anti-infectieuse), ou encore des protéines de choc thermique (HSP). Ils sont reconnus par des récepteurs spécifiques comme les récepteurs Toll-like (TLR) [5] ou les récepteurs des HSP [6, 7] (Figure 6). Des cellules présentatrices non professionnelles ne pourront maintenir l’état d’activation des lymphocytes mémoire que si elles présentent une certaine densité seuil de molécules présentatrices de peptides à leur surface, ce qui est rarement le cas des cellules tumorales. Il semble évident aujourd’hui que les signaux de danger manquent cruellement à l’activation des cellules dendritiques dans le cas du développement tumoral [4]. En effet, si les signaux de co-stimulation sont absents et la densité de peptides du CMH-I insuffisante, les CPA deviennent inductrices de tolérance (Figure 6).
Figure 4
Contrôle des cellules tumorales par le système immunitaire.
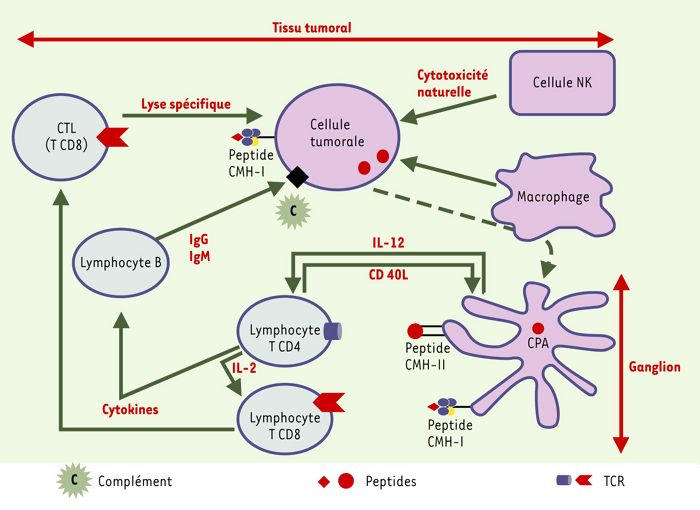
Les cellules présentatrices d’antigène (CPA) déclenchent la réponse immunitaire spécifique dans les aires T des ganglions lymphatiques. Les cellules tumorales exposent des peptides associés à des molécules HLA de classe I à leur surface. La reconnaissance de ces antigènes par des lymphocytes T CD8 spécifiques entraîne leur lyse. Les macrophages sont des cellules spécialisées dans la phagocytose, mais possèdent également, comme les cellules NK, une capacité cytotoxique naturelle. Dans un contexte « cytokinique » favorable, des lymphocytes B peuvent sécréter des anticorps capables de reconnaître les cellules tumorales et les lyser en présence de complément (ou par des mécanismes d’ADCC, antibody-dependent cellular cytotoxicity).
Comment les cellules effectrices peuvent-elles tuer les cellules tumorales ?
La lyse spécifique de la cellule tumorale par des lymphocytes T cytotoxiques est la phase effectrice d’une réponse immunitaire « idéale ». Lorsqu’elles sont activées, les cellules effectrices peuvent lyser les cellules tumorales qui présentent l’antigène. In vivo, elles doivent pour cela recirculer dans l’organisme jusqu’à leur cible. La lyse de la cellule cible se réalise par un mécanisme de cytotoxicité dépendante de la perforine et des granzymes ou d’induction d’apoptose par la fixation de la lymphotoxine ou du ligand de Fas des T CD8 sur le récepteur du TNF ou Apo1/Fas respectivement [8].
Comment les antigènes exogènes sont-ils internalisés puis présentés par la cellule dendritique ?
Dans les cellules dendritiques comme dans toute autre cellule, les vésicules d’endocytose (pinocytose ou endocytose spécifique) sont adressées au réseau des endosomes au sein duquel les éléments internalisés (antigènes exogènes) transitent (Figure 5). Dans le cas d’une tumeur, ces antigènes proviennent de débris cellulaires, de lysats ou de corps apoptotiques des cellules tumorales. Les endosomes précoces fusionnent avec des vésicules golgiennes qui favorisent une acidification progressive à l’origine de la dégradation partielle des composés endocytés. Les endosomes tardifs contenant ces éléments dégradés peuvent, dans les CPA professionnelles (comme les cellules dendritiques), fusionner avec des vésicules de rétention des molécules du complexe majeur d’histocompatibilité de classe II (CMH-II). Dans ces compartiments, les peptides endosomiques de bonne affinité se positionnent dans le sillon des molécules de classe II. Les vésicules contenant les complexes peptides CMH-II sont alors adressées à la membrane plasmique par un mécanisme d’exocytose, comme c’est le cas pour des vésicules de recyclage (Figure 5). La phagocytose est un mécanisme distinct de l’endocytose par lequel les cellules dendritiques peuvent internaliser des fragments ou encore des corps apoptotiques de cellules tumorales. Dans ce cas, et à la suite d’une reconnaissance d’éléments membranaires [9], la cellule phagocytaire encercle les particules extracellulaires. Les éléments internalisés peuvent être hydrolysés dans les lysosomes jusqu’au stade de molécules élémentaires qui pourront rejoindre les protéines cytosoliques susceptibles d’être secondairement adressées à la surface cellulaire par la voie des protéines endogènes (Figure 2). On comprend ainsi que, selon leur mode d’adressage intracellulaire, des peptides exogènes internalisés par les cellules dendritiques, peuvent se retrouver non seulement complexés à des molécules de classe II, mais également de classe I. Il s’agit du phénomène de présentation croisée ou crosspriming. Celui-ci, dans les cellules dendritiques, peut également s’expliquer par l’importance des recyclages membranaires qui ont lieu dans ce type cellulaire. En effet, les molécules de classe I naturellement ancrées dans la membrane de la cellule dendritique peuvent transiter par les endosomes au cours de ces recyclages, et les conditions physicochimiques de ce compartiment sont favorables à un échange de peptides entre molécules du CMH. Cet échange permet d’expliquer que des peptides provenant d’élements internalisés peuvent être présentés par des molécules de classe I sans passage par le réticulum endoplasmique [10]. Il faut également signaler l’importance des corps multivésiculaires d’origine endosomique et qui contiennent des particules membranaires de 60 à 80 nm de diamètre. Ces particules exposent sur leur surface des molécules du CMH de classes I et II, ainsi que des molécules de co-stimulation. Elles peuvent être exocytées et sont alors appelées exosomes.
Figure 5
Adressage des antigènes exogènes pour une présentation membranaire.
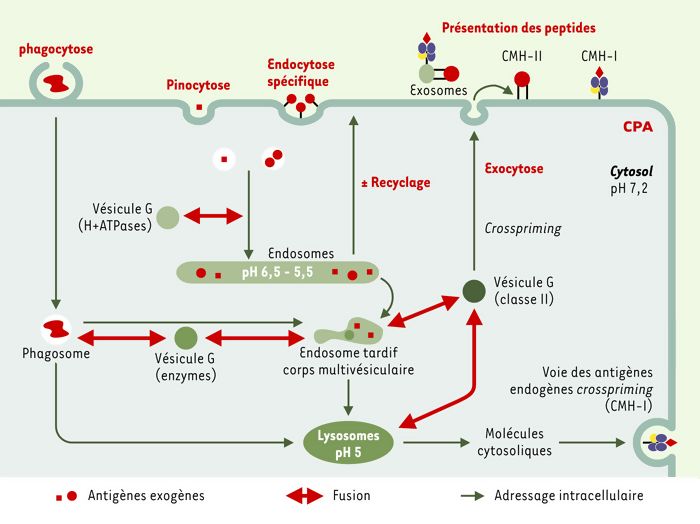
Les antigènes exogènes peuvent être internalisés par les CPA. Le réseau des endosomes est le compartiment vers lequel se dirigent les vésicules nées de la membrane plasmique par endocytose. Des bourgeonnements d’endosomes précoces fusionnent avec des vésicules golgiennes (G) qui leur apportent des ATPases à protons. Les endosomes tardifs fusionnent avec des vésicules golgiennes de rétention des molécules de classe II. Les vésicules résultantes sont adressées à la membrane plasmique comme des vésicules de recyclage ou d’exocytose. Des éléments exogènes peuvent aussi être internalisés par phagocytose. Ces deux mécanismes permettent l’adressage de molécules exogènes pour une présentation par les molécules du CMH à la surface de la CPA. La présentation de peptides endocytés par des molécules de classe I (crosspriming) est représentée.
L’induction d’une tolérance à l’égard des cellules tumorales
Chez des patients atteints de mélanome métastatique, il est fréquent de retrouver de très forts pourcentages de lymphocytes T CD8 dans le tissu tumoral. Le pourcentage de lymphocytes spécifiques d’antigènes connus, comme Melan A-MART1, par exemple, a pu être évalué [11] et il peut parfois représenter un pourcentage important des lymphocytes infiltrants. Cette spécificité est maintenant détectable grâce à l’utilisation de tétramères de molécules du CMH associées à des peptides, une méthode très sensible, dont l’utilisation est en cours d’optimisation pour caractériser l’état fonctionnel des lymphocytes spécifiques. Ainsi, chez certains patients, avec les arguments biologiques dont on dispose aujourd’hui, on serait tenté de mettre sur le compte d’une réponse immunitaire efficace l’absence de progression de la maladie. Mais, pour d’autres, malgré la présence de T CD8 dans leur tumeur, les métastases progressent alors même que ces lymphocytes, s’ils sont maintenus en culture dans des conditions favorables, se révèlent cytotoxiques à l’égard des cellules tumorales autologues. Chez ces derniers patients, les lymphocytes sont souvent localisés en périphérie tumorale, alors que, dans des mélanomes de meilleur pronostic, les lymphocytes sont réellement infiltrants. Dans ces situations in vivo, il faut admettre l’existence de mécanismes de tolérance à l’antigène qui aboutissent à l’anergie des lymphocytes spécifiques.
Figure 6
Activation des lymphocytes T spécifiques ou tolérance de l’antigène.

La faible immunogénicité des cellules tumorales peut provenir de leur faible densité membranaire en complexes peptide-CMH-I ainsi que de leur incapacité à délivrer les signaux de co-stimulation. L’absence de second signal peut induire des phénomènes d’anergie des lymphocytes T qui deviennent non répondeurs de l’antigène. À l’inverse, en présence de signaux de danger, les mêmes antigènes peuvent être adressés à des CPA activées qui les présentent efficacement. Dans ce cas, les lymphocytes T spécifiques se différencient en CTL. La connaissance de ces anomalies amène à se poser la question : comment rendre plus immunogène une cellule tumorale ?
Cette hypothèse est argumentée par des observations expérimentales. Il faut citer l’exemple de tumeurs murines (le carcinome du côlon DHOK12) dont il a été produit plusieurs variants cellulaires. Certains de ces variants peuvent être greffés à des rats syngéniques et provoquer des tumeurs métastatiques très agressives car peu immogènes : il s’agit des variants progressifs PRO.D’autres variants très immunogènes donnent au contraire chez l’animal des tumeurs qui régressent spontanément : ce sont les variants REG.Lorsqu’un animal est greffé avec des cellules PRO, un état de tolérance à la tumeur s’installe.L’immunosuppression est telle chez ces animaux qu’elle permet même la prise d’une greffe secondaire de cellules REG.à l’inverse, des rats immunisés avec des cellules REG peuvent rejeter une greffe de certains variants PRO [12]. L’utilisation de ce genre de modèles, ainsi que des observations cliniques, permettent d’arriver aujourd’hui au concept que l’état de tolérance induit par les cellules tumorales pourrait être relayé par une inaptitude des cellules présentatrices d’antigènes dans leur fonction activatrice de cellules effectrices. Des arguments sont en faveur d’un blocage de la différenciation des cellules dendritiques non seulement à proximité de la tumeur, mais également dans le sang périphérique des malades [13]. Rappelons ici encore que les cellules dendritiques immatures peuvent être tolérogènes (Figure 6).
Les mécanismes d’échappement de la cellule tumorale
La croissance de la tumeur en dépit de son antigénicité reste encore incomplètement comprise, mais de multiples mécanismes peuvent être mis en jeu dans cet échappement au contrôle par le système immunitaire. On peut dans un premier temps rappeler la faible immunogénicité des cellules tumorales qui participe au défaut d’induction ou de maintien de la réponse immunitaire. Celle-ci est en partie due à une densité de peptides à leur surface qui n’atteint pas le seuil suffisant pour une reconnaissance efficace par le TCR. On sait en effet que l’expression des molécules du CMH peut être déficiente dans les cellules tumorales. Cette déficience peut être corrigée par les IFN-α/β. La faible immunogénicité des tumeurs peut également provenir de l’absence de molécules de co-stimulation à leur surface. Les tumeurs solides n’expriment pas ou expriment peu les molécules de la famille B7, et l’absence de second signal peut induire des phénomènes d’anergie des lymphocytes spécifiques qui ne répondent plus à l’antigène [14]. Par ailleurs, contrairement aux agents infectieux, les cellules tumorales n’induisent pas de réaction inflammatoire ni de signaux de « danger », qui jouent un rôle majeur dans l’activation des cellules dendritiques [4]. Dans ce cas, la présentation des antigènes tumoraux, assimilés à des peptides du soi, est réalisée par des cellules présentatrices inductrices de tolérance. On observe alors une tolérance périphérique à l’encontre des cellules néoplasiques qui ne sont pas affectées par la présence de lymphocytes T devenus anergiques (Figure 6). Cette tolérance peut également résulter de la présence des lymphocytes T CD4 immunorégulateurs (CD4+ CD25+) qui ont été détectés parmi les TIL de tumeurs humaines et qui sont malheureusement activés par les cellules dendritiques [15] ((→) m/s 2002, n° 11, p. 1066).
Un autre phénomène s’oppose à ce que les lymphocytes spécifiques puissent mettre en place des mécanismes de défense contre les cellules tumorales : il s’agit de la sécrétion locale de facteurs suppresseurs. Des cytokines inhibitrices des réponses immunitaires peuvent être produites localement non seulement par les cellules tumorales (TGFβ, IL-10 ou PGE-2) mais également par le stroma tumoral. Cet environnement peut alors favoriser la différenciation des lymphocytes T en Th2 sécréteurs de cytokines anti-inflammatoires ou encore en lymphocytes à fonction régulatrice (Th3) [16].
Présentation des antigènes par les cellules tumorales
Pour être reconnus par des lymphocytes spécifiques, les antigènes doivent être présentés à la surface des cellules par les molécules du CMH-I. Cette présentation résulte d’un mécanisme d’adressage intracellulaire qui est commun à toutes les cellules de l’organisme. Les CMH-I présentent les peptides issus de la dégradation des protéines endogènes qui viennent de l’expression du génome de la cellule (Figure 2). Les protéines adressées aux protéasomes sont des protéines endommagées, mal configurées ou encore des protéines à dégrader dans le cadre du renouvellement programmé des protéines cellulaires. Les cellules tumorales, du fait de toutes leurs anomalies génétiques et métaboliques, engendrent des protéines anormales. Les peptides issus du protéasome, ainsi que certains petits peptides cytosoliques d’origine lysosomiale sont transportés dans la lumière du reticulum endoplasmique granuleux où ils se lient aux molécules de classe I. Pour un même antigène de tumeur, les séquences peptidiques présentées sont différentes selon le contexte HLA. Les peptides associés aux CMH-I sont ensuite véhiculés jusqu’à la surface cellulaire (Figure 2). Dans les cellules tumorales, des anomalies peuvent survenir à toutes les étapes de cet adressage intracellulaire. Des mutations portant sur les gènes du CMH-I, des transporteurs TAP, ou encore des altérations de certaines sous-unités du protéasome ont été rapportées et peuvent être responsables d’un défaut d’apprêtement des antigènes tumoraux.
Adaptation des cellules tumorales
Les cellules tumorales peuvent également être dépourvues de molécules d’adhérence aux lymphocytes telles que LFA-3 ou ICAM-1. Elles peuvent aussi exprimer des molécules anti-adhérentes telles que les mucines, échappant ainsi aux contacts avec les cellules immunocompétentes. De plus, des expériences récentes montrent que les cellules tumorales peuvent s’adapter aux défenses immunitaires. En particulier, des cellules résistantes à la lyse peuvent se trouver progressivement sélectionnées au sein des populations cellulaires tumorales [17]. Ces cellules peuvent par exemple surexprimer bcl-2, un gène anti-apoptotique. Une immunosuppression favorisée par tous ces mécanismes d’échappement peut s’installer chez les patients cancéreux au cours du développement tumoral. Il faut également rappeler qu’un excès d’antigènes peut également avoir pour conséquence un épuisement clonal par re-stimulation permanente.
Les mécanismes de défense contre les cellules tumorales demeurent encore un vrai cauchemar pour les immunologistes. Il faut bien dire que le contexte n’est pas celui de la grande success story de l’immunologie qu’ont été les vaccinations contre les maladies infectieuses [7].
Intérêt médical : des concepts au traitement des tumeurs par thérapie cellulaire
Les connaissances acquises récemment sur l’immunité antitumorale ont permis de préciser les anomalies à corriger pour atteindre une réponse immunitaire efficace. Ce que l’on attend de l’immunothérapie des cancers est l’éradication des cellules néoplasiques sans affecter les cellules normales, et cela quelle que soit leur localisation, notamment dans les métastases. L’idée d’amplifier sur le plan qualitatif et quantitatif, in vitro ou in vivo, les effecteurs de la réponse immunitaire antitumorale est le concept de base. Il est maintenant décliné avec tous les partenaires de la réponse. Les stratégies thérapeutiques sont variées. Elles ont toutes pour objectif de lever l’anergie des cellules potentiellement réactives en les stimulant dans des conditions optimisées. Le traitement des tumeurs par immunisation peut être actif ou passif, selon qu’il s’agit d’induire in vivo chez le malade la réponse immunitaire ou bien d’apporter au patient des effecteurs cytotoxiques de « suppléance ». Parmi ces traitements, certains ont fait leurs preuves. C’est le cas de l’injection de cytokines, comme par exemple l’IL-2 et l’IFN-α qui sont devenues le traitement de référence dans les cancers du rein métastatiques [18]. D’autres sont en cours d’évaluation clinique. C’est le cas des thérapies cellulaires ainsi que des vaccinations contre le cancer qui suscitent de réels espoirs car les résultats expérimentaux obtenus à ce jour ont été très significatifs.
Thérapies cellulaires par effecteurs cytotoxiques
Il faut rappeler les expériences pionnières de l’équipe de Rosenberg qui, dès 1984 [19], obtenait avec des injections de lymphocytes activés une réduction du nombre et de la taille des métastases dans plusieurs modèles de tumeur murine. Des injections d’IL-2 potentialisent l’efficacité de ces cellules. Dans le modèle des macrométastases de l’adénocarcinome du côlon MC 38, c’est non seulement une disparition des métastases hépatiques ou pulmonaires qui était observée, mais une guérison de 100 % des souris [20]. Les souris guéries par ce traitement étaient de plus immunisées contre une nouvelle greffe des cellules provenant de la tumeur d’origine, car les lymphocytes transférés déclenchaient in vivo une cascade de sécrétions de cytokines et de coopérations cellulaires amplifiant la réponse spécifique antitumorale. Les résultats observés sur les tumeurs murines ayant été confirmés par plusieurs équipes, des essais cliniques ont été envisagés dans le traitement du mélanome et du cancer du rein. Après de nombreuses études de faisabilité, il est apparu que des lymphocytes T CD8 présentant toutes les caractéristiques fonctionnelles attendues pour espérer une efficacité clinique pouvaient être amplifiés à partir de pièces opératoires. Beaucoup d’efforts ont été focalisés sur les procédés techniques de leur amplification. Aucun de ces protocoles n’est pleinement satisfaisant car la fréquence des lymphocytes T spécifiques des cellules tumorales demeure extrêmement variable d’un patient à l’autre. Le travail de notre groupe a pour objectif de sélectionner des lymphocytes en utilisant des cellules dendritiques chargées[*] avec les antigènes de la tumeur autologue [21].
Certaines équipes ont mené des essais cliniques avec des TIL, malgré les imperfections des protocoles. Dans les cancers du rein métastasés, ces essais ont donné des réponses complètes dans plusieurs études et des réponses partielles dans toutes les études. Par exemple, plusieurs réponses complètes ont été rapportées dans les essais menés par Belldegrun en 1993 puis en 1997. Dans un essai de phase II, la moitié des patients a reçu des suspensions cellulaires enrichies en T CD8 (plusieurs milliards de cellules), et c’est dans ce groupe que le plus grand nombre de réponses a été observé [22]. Des essais cliniques de traitement des mélanomes par des lymphocytes activés ont également été menés en France. Un essai vient de se terminer à Nantes (Inserm U.463, Nantes, France) [23] qui montre une augmentation significative de la survie sans rechute des patients traités par des lymphocytes activés, par rapport au groupe témoin. Une corrélation est observée entre l’efficacité clinique et le pourcentage de lymphocytes T spécifiques des antigènes de tumeur. Ces résultats sont porteurs d’espoir.
Immunothérapie active non spécifique
À côté des méthodes d’immunothérapie adoptive, où les cellules immunocompétentes sont injectées « toutes faites » au malade, se positionnent les méthodes d’immunothérapie active du cancer. Si l’intérêt des chercheurs se focalise aujourd’hui surtout sur les méthodes d’immunothérapie active spécifique, il faut cependant rappeler que l’immunothérapie active non spécifique, fondée sur le principe d’une stimulation globale du système immunitaire chez des patients cancéreux, a déjà fait ses preuves : l’injection intralésionnelle de BCG est une méthode de référence dans le traitement des tumeurs non invasives de la vessie.
Immunothérapie active spécifique : vaccinations avec ou sans thérapie cellulaire par cellules présentatrices ?
Pour produire in vivo chez les malades des cellules effectrices capables de lyser spécifiquement les cellules tumorales, les antigènes tumoraux doivent être présentés de nouveau à l’organisme dans des conditions favorables au déclenchement d’une réponse immunitaire. C’est ce qui a été tenté expérimentalement dans les essais d’injection de cellules tumorales modifiées, par exemple en les transfectant avec des ADNc codant pour des molécules de co-stimulation (B7) ou pour des cytokines (IL-2). L’augmentation de leur immunogénicité induit une protection anti-tumorale chez l’animal. C’est le même objectif qui est poursuivi dans les essais d’immunisation avec de simples peptides-antigènes de tumeur. Des essais cliniques menés en utilisant les peptides MAGE ont démontré des régressions de métastases dans des mélanomes. Dans le cas des antigènes de différenciation, l’apparition d’un vitiligo est souvent associée à une régression tumorale du mélanome. Actuellement, des essais sont menés avec des antigènes de tumeur qui ont été fusionnés avec des ligands de la surface des cellules dendritiques, comme des chimiokines ou des fragments Fc d’immunoglobulines par exemple, pour adresser cet antigène à la cellule dendritique comme un missile vers sa cible [24]. De la même manière, des vecteurs viraux ou de l’ADN nu porteurs du gène codant pour l’antigène de tumeur sont en évaluation et pourraient constituer de véritables vaccins contre certains types de cancers [24].
Les essais d’injection de cellules présentatrices d’antigène ont aussi pour objectif la stimulation des défenses anti-tumorales. Ils sont en pleine évolution et, chaque année, de nouvelles statégies, originales et ingénieuses, donnent des résultats expérimentaux qui suscitent beaucoup d’enthousiasme. Les cellules dendritiques peuvent être chargées avec des peptides tumoraux, des extraits de cellules tumorales, des corps apoptotiques, des exosomes, ou encore être fusionnées avec des cellules tumorales. Dans toutes ces situations expérimentales, elles possèdent la capacité d’activer in vitro des lymphocytes qui acquièrent des propriétés cytotoxiques. Des essais précliniques montrent que les cellules dendritiques présentent un pouvoir curatif et préventif à l’égard de tumeurs greffées. Dans des modèles murins de tumeur gliale [25], de mastocytome [26, 27] ou de mélanome [28]. l’injection sous-cutanée de cellules dendritiques chargées en antigènes de tumeur entraîne une infiltration de la tumeur par des CTL, associée à un effet thérapeutique.
Les premiers essais cliniques ont été entrepris par l’équipe de G. Murphy. Il s’agissait de patients exprimant le CMH de classe I HLA-A2, atteints de cancer de la prostate métastasé, et les peptides choisis provenaient de la molécule PSMA. Des réponses partielles (9/33) et un effet significatif sur la survie ont été obtenus dans ces essais [29]. Les résultats de l’essai de traitement de mélanomes métastatiques mené par le groupe de F.O. Nestlé [30] méritent également d’être soulignés. Les cellules dendritiques étaient chargées avec des peptides caractéristiques des haplotypes HLA-A2 ou HLA-A1 ainsi qu’avec des lysats de la tumeur autologue chez quatre patients. Sur les 16 patients inclus, 5 réponses ont été obtenues dont deux avec les lysats tumoraux [30]. L’utilisation de lysats tumoraux présente l’intérêt majeur d’être a priori applicable à toutes les tumeurs, même si les antigènes de ces tumeurs ne sont pas identifiés et sans nécessité pour le patient d’appartenir à un groupe HLA particulier. Plusieurs approches expérimentales, comme la fusion de cellules dendritiques avec des liposomes ou encore leur transfection avec des ARN tumoraux sont encore actuellement menés pour améliorer la présentation des antigènes de tumeur par les molécules de classe I. Une façon efficace d’obtenir l’expression endogène d’antigènes de tumeurs par les cellules dendritiques est de les fusionner avec les cellules tumorales. Un essai clinique vient d’en faire la preuve. Dix-sept patients atteints de cancer du rein métastatique ont été vaccinés avec 100 millions de cellules résultant de l’électrofusion de cellules dendritiques allogéniques avec des cellules tumorales autologues. Quatre patients ont montré un rejet complet de toutes leurs métastases. Trois autres patients ont bénéficié d’une réduction de plus de 50 % de tous les sites tumoraux pendant plus de 3 mois [31]. Les analyses histologiques et biologiques confirment une réponse immunitaire anti-tumorale relayée par des lymphocytes T CD8 spécifiques. Ce résultat est supérieur à ce qui est obtenu avec les traitements habituels du cancer du rein métastatique[**].
Une explosion d’essais cliniques
Depuis la découverte des premiers antigènes de tumeur, plus de 80 peptides antigéniques ont été caractérisés. L’étape de la démonstration de leur utilisation en thérapeutique est en cours. Actuellement, une centaine d’essais cliniques de thérapie vaccinale du cancer sont référencés par le NCI (National Cancer Institute, USA) (http:// cancernet.nci.nih.gov). Certains de ces essais utilisent des peptides, seuls ou en association, d’autres utilisent des extraits de tumeur autologue ou de lignées cellulaires « sécurisées »[***]. Les antigènes sont, selon les essais, utilisés directement ou après chargement sur des CPA, ou encore en combinaison avec des injections de cellules mononucléées ou de cytokines. Des essais sont menés dans pratiquement tous les types de tumeurs. Il sprincipalement d’essais de phase I ou II, encore souvent menés dans des contextes HLA particuliers. D’après les résultats préliminaires annoncés dans différents colloques, il apparait que la thérapie cellulaire par CPA améliore les résultats obtenus avec les seules vaccinations par peptides antigéniques. D’une façon générale, les réponses thérapeutiques à une immunothérapie sont meilleures lorsque celle-ci est proposée à un stade précoce de la maladie. Les questions qui devront maintenant être abordées concernent l’optimisation de la maturation des cellules dendritiques à usage thérapeutique, la standardisation des protocoles de leur préparation, les doses, les fréquences ainsi que la voie d’injection les plus favorables. Pour conclure, il faut rappeler que le choix de la statégie d’immunothérapie (active ou passive) devra tenir compte de l’état d’immunosuppression du patient qui est souvent corrélé au stade de sa maladie. Il est très probable que, à l’avenir, plusieurs approches complémentaires devront être combinées [32], comme cela a été le cas avec la chimiothérapie des cancers.
Appendices
Notes
-
[*]
Des cellules dendritiques chargées sont des cellules qui ont été incubées avec un antigène (ou son précurseur) pendant une période suffisante pour devenir cellule présentatrice de cet antigène.
-
[**]
Voir la controverse récente publiée dans Science au sujet de cet essai (Science 2002 ; 298 : 1531).
-
[***]
Une lignée sécurisée est une lignée cellulaire sans potentiel infectieux.Son usage dans le cadre d'un essai thérapeutique doit être autorisé par l'AFSSAPS (Agence française de sécurité sanitaire des produits de santé).
Références
- 1. Boon T, Brichard VG, Eynde BVD. Antigènes de rejet des tumeurs et immunothérapie spécifique du cancer. Med Sci 1995 ; 11 : 1279-87.
- 2. Eynde BJVD, Gaugler B, Probst-Kepper M, et al. A new antigen recognized by cytolytic T lymphocytes on a human kidney tumor results from reverse strand transcription. J Exp Med 1999 ; 190 : 1793-800.
- 3. Rodriguez A, Regnault A, Kleijmeer M, Ricciardi-Castagnoli P, Amigorena S. Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat Cell Biol 1999 ; 1 : 362-8.
- 4. Matzinger P. The danger model : a renewed sense of self. Science 2002 ; 296 : 301-5.
- 5. Imler J, Hoffman J. Toll receptors in innate immunity. Trends Cell Biol 2001 ; 11 : 304-11.
- 6. Delneste Y, Magistrelli G, Gauchat J, et al. Involvement of Lox-1 in dendritic cell mediated antigen cross-presentation. Immunity 2002 ; 17 : 353-62.
- 7. Banchereau J, Schuler-Thurner B, Palucka AK, Schuler G. Dendritic cells as vectors for therapy. Cell 2001 ; 106 : 271-4.
- 8. Barry M, Bleackley RC. Cytotoxic lymphocytes : all roads lead to death. Nat Immunol 2002 ; 2 : 401-9.
- 9. Tjelle TE, Lovdal T, Berg T. Phagosome dybnamics and function. Bioessays 2000 ; 22 : 255-63.
- 10. Grommé M, Uytdehaag FG, Janssen H, et al. Recycling MHC class I molecules and endosomal peptide loading. Proc Natl Acad Sci USA 1999 ; 96 : 10326-31.
- 11. Pittet MJ, Speiser DE, Valmori D, et al. Ex vivo analysis of tumor antigen specific CD8+ T cell responses using MHC/peptide tetramers in cancer patients. Int Immunolpharmacol 2001 ; 1 : 1235-47.
- 12. Bonotte B, Favre N, Moutet M, et al. Bcl2-mediated inhibition of apoptosis prevents immunogenicity and restores tumorigenicity of spontaneously regressive tumors. J Immunol 1998 ; 161 : 1433-8.
- 13. Almand B, Clark JI, Nikitina E, et al. Increased production of immature myeloid cells in cancer patients : a mechanism of immunosuppression in cancer. J Immunol 2001 ; 166 : 678-89.
- 14. Pardoll DM. Cancer vaccines. Nat Med 1998 ; 4 (suppl) : 525-31.
- 15. Sakaguchi S. Regulatory T cells : key controllers of immunologic self-tolerance. Cell 2000 ; 101 : 455-8.
- 16. Chouaib S, Asselin-Paturel C, Mami-Chouaib F, Caignard A, Blay JY. The host-tumor immune conflict : from immunosuppression to resistance and destruction. Immunol Today 1997 ; 18 : 493-7.
- 17. Bodey B, Bodey B Jr, Siegel SE, Kaiser HE. Failure of cancer vaccines : the significant limitations of this approach to immunotherapy. Anticancer Res 2000 ; 20 : 2665-76.
- 18. Negrier S, Escudier B, Lasset C, et al. Recombinant human interleukin-2, recombinant human interferon alpha-2a, or both in metastatic renal-cell carcinoma. N Engl J Med 1998 ; 338 : 1272-8.
- 19. Mulé JJ, Shu S, Schwarz SL, Rosenberg SA. Adoptive immunotherapy of established pulmonary metastases with LAK cells and recombinant interleukin-2. Science 1984 ; 225 : 1487-9.
- 20. Rosenberg S, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 1986 ; 233 : 1318-21.
- 21. Bouet-Toussaint F, Genetet N, Rioux-Leclercq N, et al. Interleukin 2 expanded lymphocytes from lymph node and tumor biopsies of human renal cell carcinoma, breast and ovarian cancer. Eur Cytokine Netw 2000 ; 11 : 217-24.
- 22. Figlin R, Pierce W, Kaboo R, et al. Treatment of metastatic renal cell carcinoma with nephrectomy, interleukin-2 and cytokine-primed or CD8+ selected tumor infiltrating lymphocytes from primary tumor. J Urol 1997 ; 158 : 740-5.
- 23. Dreno B, Nguyen JM, Khammari A, et al. Randomized trial of adoptive transfer of melanoma tumor-infiltrating lymphocytes as adjuvant therapy for stage III melanoma. Cancer Immunol Immunother 2002 ; 51 : 539-46.
- 24. Biragyn A, Kwak LW. Designer cancer vaccines are still in fashion. Nat Med 2000 ; 6 : 966-8.
- 25. Liau LM, Black KL, Prins RM, et al. Treatment of intracranial gliomas with bone marrow-derived dendritic cells pulsed with tumor antigens. J Neurosurg 1999 ; 90 : 1115-24.
- 26. Zitvogel L, Mayodormo J, Tjandrawan T, et al. Therapy of murine tumors with tumor peptide-pulsed dendritic celles : dependence on T cells, B7 costimulation, and T helper cell-1 associated cytokines. J Exp Med 1996 ; 183 : 87-97.
- 27. Lespagnard L, Mettens P, Verheyden AM, et al. Dendritic cells fused with mastocytoma cells elicit therapeutic antitumor immunity. Int J Cancer 1998 ; 13 : 250-8.
- 28. Wang J, Saffold S, Cao X, Krauss J, Chen W. Eliciting T cell immunity against poorly immunogenic tumors by immunization with dendritic cell-tumor fusion vaccines. J Immunol 1998 ; 161 : 5516-24.
- 29. Murphy G, Tjoa B, Ragde H, et al. Phase I clinical trial : T-cell therapy for prostate cancer using autologous dendritic cells pulsed with HLA-A0201-specific peptides from prostate-specific membrane antigen. Prostate 1996 ; 29 : 371-80.
- 30. Nestlé FO, Alijagic S, Gilliet M, et al. 1998. Vaccination of melanoma patients with peptide or tumor lysate-pulsed dendritic cells. Nat Med 1998 ; 4 : 328-32.
- 31. Kugler A, Stuhler G, Walden P, et al. Regression of human metastatic renal cell carcinoma after vaccination with tumor cell-dendritic cell hybrids. Nat Med 2000 ; 6 : 332-6.
- 32. Dudley ME, Wunderlich JR, Yang JC, et al. A phase I study of non myeloablative chemotherapy and adoptive transfer of autologous tumor-antigen specific T lymphocytes in patients with metastatic melanoma. J Immunother 2002 ; 25 : 243-51.
List of figures
Figure 1
Obtention de clones CTL antitumoraux autologues.
L’expérience de clonage des lymphocytes T spécifiques des antigènes tumoraux a consisté à cultiver des cellules tumorales provenant d’une biopsie, à les irradier, puis à les mélanger à des lymphocytes circulants du même patient. Les lymphocytes dirigés contre un antigène tumoral prolifèrent et se différencient. Ils sont alors clonés et testés pour leur capacité de lyser certaines cellules tumorales issues de la culture primaire. Des sous-populations tumorales peuvent ainsi être sélectionnées et les gènes codant pour ces antigènes dits « de tumeur » sont identifiés par comparaison des génomes des différentes sous-populations (d’après [1]).
Figure 2
Adressage des antigènes endogènes pour une présentation membranaire.
Les peptides issus du protéasome, ainsi que certains petits peptides cytosoliques d’origine lysosomiale, sont transportés dans la lumière du réticulum endoplasmique granuleux (REG) par un complexe de transport TAP (transporter-associated with antigen processing). Les molécules de classe I synthétisées dans le REG accueillent les peptides qui y sont transférés. Cette association est complétée par la fixation de la β2-microglobuline (β2-m, en jaune) et est chaperonnée par des protéines d’adressage (HSP). Les peptides associés aux molécules CMH-I sont ensuite véhiculés dans des vésicules recouvertes de coatomères (manteau impliqué dans la formation des vésicules adressées au réticulum andoplasmique) qui empruntent le flux vectoriel permanent. Ils se positionnent à la surface cellulaire par un mécanisme d’exocytose constitutive.
Figure 3
Reconnaissance spécifique et activation du lymphocyte T par une cellule présentatrice d’antigène (CPA).
L’antigène présenté par les molécules du CMH (de classe II ou I) est reconnu par des lymphocytes T (CD4 ou CD8 respectivement) spécifiques de cet antigène. Les lymphocytes ne seront activés que si le signal (1) (reconnaissance spécifique du peptide) est accompagné des signaux dits de co-stimulation (2). Le second signal est délivré au lymphocyte T lors de l’interaction de récepteurs accessoires avec des ligands situés à la surface des CPA : les molécules B7 jouent ce rôle par leur liaison aux récepteurs CD28. En l’absence de ce signal 2, le lymphocyte devient tolérant à l’antigène. Différentes molécules d’adhérence stabilisent ces interactions.
Figure 4
Contrôle des cellules tumorales par le système immunitaire.
Les cellules présentatrices d’antigène (CPA) déclenchent la réponse immunitaire spécifique dans les aires T des ganglions lymphatiques. Les cellules tumorales exposent des peptides associés à des molécules HLA de classe I à leur surface. La reconnaissance de ces antigènes par des lymphocytes T CD8 spécifiques entraîne leur lyse. Les macrophages sont des cellules spécialisées dans la phagocytose, mais possèdent également, comme les cellules NK, une capacité cytotoxique naturelle. Dans un contexte « cytokinique » favorable, des lymphocytes B peuvent sécréter des anticorps capables de reconnaître les cellules tumorales et les lyser en présence de complément (ou par des mécanismes d’ADCC, antibody-dependent cellular cytotoxicity).
Figure 5
Adressage des antigènes exogènes pour une présentation membranaire.
Les antigènes exogènes peuvent être internalisés par les CPA. Le réseau des endosomes est le compartiment vers lequel se dirigent les vésicules nées de la membrane plasmique par endocytose. Des bourgeonnements d’endosomes précoces fusionnent avec des vésicules golgiennes (G) qui leur apportent des ATPases à protons. Les endosomes tardifs fusionnent avec des vésicules golgiennes de rétention des molécules de classe II. Les vésicules résultantes sont adressées à la membrane plasmique comme des vésicules de recyclage ou d’exocytose. Des éléments exogènes peuvent aussi être internalisés par phagocytose. Ces deux mécanismes permettent l’adressage de molécules exogènes pour une présentation par les molécules du CMH à la surface de la CPA. La présentation de peptides endocytés par des molécules de classe I (crosspriming) est représentée.
Figure 6
Activation des lymphocytes T spécifiques ou tolérance de l’antigène.
La faible immunogénicité des cellules tumorales peut provenir de leur faible densité membranaire en complexes peptide-CMH-I ainsi que de leur incapacité à délivrer les signaux de co-stimulation. L’absence de second signal peut induire des phénomènes d’anergie des lymphocytes T qui deviennent non répondeurs de l’antigène. À l’inverse, en présence de signaux de danger, les mêmes antigènes peuvent être adressés à des CPA activées qui les présentent efficacement. Dans ce cas, les lymphocytes T spécifiques se différencient en CTL. La connaissance de ces anomalies amène à se poser la question : comment rendre plus immunogène une cellule tumorale ?