Abstracts
Résumé
Accompagnant depuis la nuit des temps la vie des hommes, le stress n’apparaît pas sans conséquences sur leur état de santé. Le rôle des peurs, des émotions et des contraintes prégnantes dans l’apparition de maladies organiques ou psychiques a, depuis longtemps, fait l’objet d’observations cliniques. Les progrès récents des neurosciences et l’identification de mécanismes moléculaires communs aux systèmes nerveux et immunitaire offrent des pistes de recherches ou, du moins, indiquent des directions à prendre. En particulier celles des voies biologiques communes impliquées dans différents types de stress, psychologique, traumatique, endotoxique ou infectieux, dont les effets peuvent se conjuguer et se renforcer. Nous avons fait le choix des cytokines, molécules par excellence de l’immunité, mais aussi présentes dans le cerveau ainsi que leurs récepteurs, pour illustrer l’existence de supports moléculaires aux interactions entre stress, immunité et maladies mentales. Ce thème, déjà abordé par d’autres, est généralement traité par des neurobiologistes pour des neurobiologistes. Nous avons souhaité que cet article s’adresse plus particulièrement à ceux, médecins, psychiatres ou étudiants en psychologie et d’autres pour qui le monde des molécules paraît bien éloigné du psychisme.
Summary
Stress, immunity and nervous system physiology have long been viewed as closely interwoven. Ancient clinical observations recount that fears, emotions and permanent constreints have consequences on both the physical and psychological health status of individuals.
Recent advances in the understanding of neurobiological mechanisms and the identification of molecular signaling pathways common to the nervous and the immune systems offer new avenues for present and future research. In this review, we have chosen the cytokine family to illustrate these interactions. A short historical view is presented and time questioning of clinicians on how far molecules may be involved in these interactions is addressed to neurobiologists. This is an attempt to built a bridge between neurobiologists and clinicians, especially those who, in the field of psychology and mental disorders, wonder whether molecules have something to do with the mind.
Article body
La question des rapports entre le stress, le traumatisme, l’immunité et les troubles psychiques est déjà ancienne en psychiatrie [1]. Ces rapports ont été initialement envisagés, à partir d’arguments cliniques et thérapeutiques. Ainsi, différents neuropsychiatres ont mentionné l’existence des maladies infectieuses dans les antécédents des troubles mentaux, certains évoquant plutôt un mécanisme post-encéphalitique, d’autres un mécanisme allergique. D’autres encore ont signalé la coexistence de troubles mentaux chez des personnes atteintes d’affections immunitaires (polyarthrite, lupus, asthme), et réciproquement. Le rôle de l’émotion dans le déclenchement de processus infectieux et immunitaires, ainsi que dans l’apparition et l’évolution de troubles psychiques, est également apparu très tôt. Ces différentes observations avaient conduit les psychiatres à utiliser, avant la découverte des psychotropes actuels, les traitements de choc, anti-allergiques et anti-infectieux [2].
Depuis une dizaine d’années, de nombreux travaux portent sur les effets immunologiques et neurobiologiques du stress psychologique en fonction de ses caractères objectif et subjectif, de sa durée, de la capacité ou non du sujet d’y faire face, de son caractère interpersonnel ou non, ainsi que du contexte dans lequel il s’inscrit [3]. Parmi les facteurs de risque qui prédisposent certains sujets à la survenue de troubles, l’importance d’un contexte de traumatisme antérieur et d’insécurité de la première enfance, voire de stress pré-natal est soulignée [4], [5]. Ces observations cliniques [6] et les investigations qui les accompagnent trouvent aujourd’hui un prolongement dans des constatations expérimentales recueillies au niveau neurobiologique. Les interactions entre le système nerveux central (SNC), le stress et le système immunitaire sont mieux connues, en particulier le rôle qu’y jouent les cytokines sur les cellules ainsi que comme acteurs de liaison et de médiation entre les deux systèmes. Il apparaît non seulement que ces agents peuvent être mobilisés au niveau biologique par des stress apriori très hétérogènes (psychiques, infectieux, toxiques, traumatiques), mais qu’ils peuvent intervenir comme de véritables neuromédiateurs, avoir une expression cliniquement détectable sous la forme de sensations psychiques (fatigue, sommeil…), voire de comportements adaptatifs (« de maladie »), participer à des activités mentales telles que la mémoire, mais aussi - à des concentrations extra-physiologiques - au déclenchement d’authentiques dépressions. Certes, ces données n’éclairent que très partiellement la façon dont le psychisme et l’immunité, dans leurs acceptions les plus larges, peuvent interagir. Elles donnent néanmoins une base concrète à la façon dont certains processus biologiques, mentaux et affectifs peuvent être associés. Des ébauches de schémas d’enchaînements et de processus, où les implications immunitaires du stress ne peuvent être ignorées, se dessinent et permettent d’identifier des pistes de recherche. Cet article tente d’y contribuer en mettant en relation quatre modalités d’implication des cytokines dans le cerveau dans différents cas: dans la réponse biologique au stress au niveau de l’axe hypothalamo-hypophyso-surrénalien, face aux hyperactivités et aux agressions du SNC, dans la modulation de différentes activités cérébrales et mentales et dans le déclenchement de certaines maladies mentales.
Cytokines et réponse biologique au stress au niveau de l’axe hypothalamo-hypophyso-surrénalien
Le stress est associé à une activation de plusieurs systèmes neuroendocriniens, incluant le système nerveux sympathique et l’axe hypothalamo-hypophyso-surrénalien (HPA selon la terminologie anglo-américaine) (Figure 1). L’ensemble constitue une boucle de régulation sensible en de nombreux points aux activités nerveuses et immunitaires, elles-mêmes soumises aux facteurs d’environnement.
Figure 1
Représentation schématique des interventions des cytokines dans le fonctionnement de l’axe du stress.
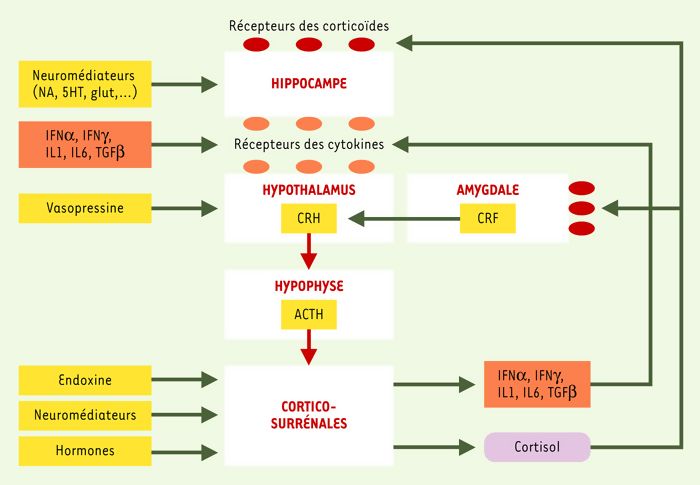
Les neurones parvocellulaires du noyau paraventriculaire de l’hypothalamus produisent la CRH, hormone également abondante dans l’amygdale. Cet autre site joue un rôle crucial dans la perception de peur et d’anxiété et la production de noradrénaline qui stimule les neurones du noyau paraventriculaire de l’hypothalamus. Dans des conditions normales, l’action de la CRH sur les cellules corticotrophiques de l’hypophyse induit et règle la sécrétion de l’ACTH, soit en l’augmentant à la suite d’un stress, soit en la réduisant en fonction de la concentration élevée de corticoïdes. Les effets de la CRH sont eux-mêmes modulés par la vasopressine et par certaines cytokines. Véhiculé par la circulation sanguine, l’ACTH agit sur les corticosurrénales qui, en réponse, produisent le cortisol. Ce dernier, utilisant également la voie sanguine, agit notamment sur l’hypothalamus, sur l’hippocampe et sur l’amygdale et participe à la régulation de l’immunité.
L’axe HPA a été abondamment étudié et décrit [7]. Une des particularités des acteurs impliqués est d’avoir à la fois des effets d’information et de régulation dans des processus biologiques en cascade, ainsi que des effets propres perceptibles sur le psychisme (corticotropin-releasing hormone ou CRH, cortisol, cytokines, sérotonine) qui peuvent eux-mêmes être à l’origine de comportements secondaires potentiellement pathologiques. Certains d’entre eux peuvent également provoquer une altération du système nerveux ou la réduire. Riches en récepteurs des corticoïdes, les neurones hippocampiques peuvent ainsi subir un processus dégénératif sous l’effet d’une concentration locale excessive de glucocorticoïdes [8]. Celle-ci peut engendrer une toxicité par la libération accrue des acides aminés excitateurs, elle-même amplifiée par l’augmentation de la densité de leurs récepteurs liée à cette concentration excessive. Les pertes neuronales touchent principalement les neurones pyramidaux des aires CA3 et CA1 [9]. Or, l’hippocampe est la région-clé du cerveau pour les processus mentaux d’apprentissage et de mémorisation. L’amygdale, fortement impliquée dans l’émotivité, est riche en récepteurs stéroïdiens dont l’activation accroît, elle aussi, la libération de CRH [10].
Les cytokines, polypeptides médiateurs de l’immunité et de l’inflammation, participent à des fonctions biologiques très diverses dans plusieurs systèmes, dont le SNC. La présence de cytokines inflammatoires dans le cerveau a été initialement attribuée à une synthèse exclusive par les cellules myélo-monocytaires infiltrantes, puisque les cellules nerveuses n’étaient pas supposées produire et utiliser ces molécules. Puis des études sur des cellules en culture ont montré une capacité de synthèse du TNFα par les cellules gliales, les astrocytes et surtout par la microglie [11]. L’expression dans le cerveau des cytokines peut se produire sous la forme d’un processus en chaîne. Ainsi, le TNFα stimule la production de CSF-1 in vitro par les astrocytes. Le CSF-1 induit la prolifération de la microglie. La microglie activée est elle même capable de libérer du TNFα et de l’interleukine-1 (IL-1). Le TNFα induit une prolifération des astrocytes.
Les récepteurs de différentes cytokines sont présents dans le cerveau, soit dans l’hypothalamus même, soit dans des structures cérébrales directement connectées à l’hypothalamus. Ceux de l’IL-1 sont situés principalement dans l’hippocampe et l’hypothalamus (revue dans [12]); ceux de l’IL-6 ont été localisés dans l’hippocampe, l’habenulae, l’hypothalamus, le gyrus dentelé et le cortex piriforme [12]. L’expression du récepteur de l’IL-6 augmente considérablement dans l’hypothalamus au moment où s’achève la maturation de l’axe HPA. Des récepteurs du TNF sont exprimés sur diverses populations de cellules neuronales dans de nombreuses régions du cerveau dont l’hippocampe, le cortex, l’amygdale et les noyaux de la base [12], ainsi que sur les oligodendrocytes [13]. Ces observations renforcent sérieusement l’hypothèse d’un pool de TNFα modulant des activités neuronales. En effet, neurones et cellules gliales du cerveau, étant capables de synthétiser le TNFα ainsi que son récepteur, peuvent utiliser cette molécule dans le cadre d’un système de communication neuro-neuronal ou glio-neuronal.
De nombreux effets du TNFα sur le système nerveux pourraient être expliqués par son action sur la voie de signalisation par les protéines G [14], voie activée après la stimulation de nombreux récepteurs des neuromédiateurs. Le TNFα pourrait avoir accès aux éléments cytoplasmiques des neurones par internalisation du complexe récepteur-ligand, de nombreuses populations neuronales exprimant l’un ou l’autre des récepteurs de cette cytokine.
L’axe HPA est sensible à l’action complexe des cytokines, qu’elles soient produites dans le SNC ou acheminées par voie sanguine du fait de la perméabilité de la barrière hémato-encéphalique dans la région hypothalamique. L’IL-1, l’IL-6, l’interféron et le TNFα ont une action activatrice sur cet axe. La libération par les surrénales de glucocorticoïdes inhibe la réponse inflammatoire et permet de contenir la réaction immunitaire.
Le noyau paraventriculaire de l’hypothalamus (NPV), où sont localisés les neurones producteurs de CRH, est sensible à l’action de l’IL-1 et à celle de son antagoniste, l’IL-1Ra [15]. La libération de CRH, est stimulée par les cytokines pro-inflammatoires IL-1β, IL-6 et l’IFNα, mais inhibée par la cytokine anti-inflammatoire TGFα.
L’activation de l’axe HPA par les cytokines s’accompagne également d’effets sur le métabolisme de certains neuromédiateurs. Ainsi l’administration d’IL-1 stimule le métabolisme de la noradrénaline et de la sérotonine, notamment dans l’hypothalamus. Ces effets sont semblables à ceux qui sont obtenus par administration du LPS, ou du virus de l’influenza [16].
Les surrénales constituent un autre site d’interaction entre l’axe HPA et les cytokines [17]. En effet, la production des principales cytokines pro- et anti-inflammatoires se situe dans le cortex des surrénales. Elle y est réglée par de nombreux agents moléculaires, dont les endotoxines, les neuromédiateurs et certaines hormones, dont les glucocorticoïdes.
Les cytokines face aux hyperactivités et aux agressions du SNC
Différentes activités modulatrices des cytokines sur les neurones ont été répertoriées [12] (Figure 2).
Figure 2
Représentation schématique des relations entre cytokines intra-cérébrales et pathologies nerveuses et/ou psychiques.
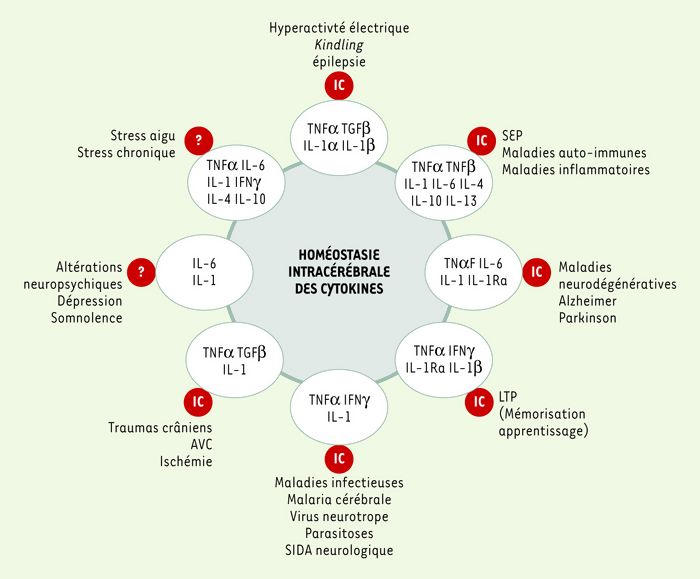
En regard de chaque groupe de pathologies figure la liste des cytokines impliquée dans une ou plusieurs de ces pathologies. Les lettres IC dans les ronds rouges indiquent que la présence des cytokines dans le cerveau a été démontrée. Le point d’interrogation indique une absence de démonstration de cette présence. Les références sont données dans le texte ou, à défaut, citées dans [12].
Neuroprotection et adaptation du système nerveux aux hyperactivités neurotoxiques
Des actions de l’IL-6 favorables à la survie et à la différenciation de neurones en culture ont été décrites. L’IL-6 et son ARN messager apparaissent dans le système nerveux lors de dégénérescences et exercent un rôle neuroprotecteur [18]. Suggérée depuis quelques années par de rares et intéressants travaux signalant, par exemple, que des crises audiogènes induisent une synthèse d’IL-1β dans le cerveau, cette idée a gagné en crédibilité à la suite de travaux récents montrant, à l’aide d’un modèle de kindling chez le rat, une forte induction de l’expression d’IL-1β, de TNFα et de TGFα1 dans le cerveau. De façon intéressante, ces activations concernaient non seulement l’amygdale, site de déclenchement du kindling, mais aussi l’hippocampe et le cortex, régions habituellement concernées par les décharges épileptiques [19].
Aggravation des processus neurodégénératifs
De nombreux travaux témoignent de l’implication des cytokines pro-inflammatoires dans la neurodégénérescence [12], en dépit de l’absence d’effet délétère direct de ces cytokines sur des cultures de neurones sains.
Expression dans le cerveau au cours d’infections, de traumatismes crâniens et de pathologies
Au cours de l’infection du SNC par le virus VIH-1, la concentration des cytokines dans le LCR est élevée. De fortes expressions du TNFα et de l’IL-1 sont observées dans les cellules microgliales et sur les cellules endothéliales respectivement. L’évolution du SIDA vers la démence s’accompagne d’une augmentation significative du TNFα dans le parenchyme cérébral des malades [20].
D’autres maladies s’accompagnent d’une augmentation, parfois considérable, de la concentration sérique en TNFα, telle la malaria cérébrale, les parasitoses et les traumatismes crâniens [21]. L’infection par le trypanosome induit chez le rat une expression intracérébrale importante et durable d’IL-1α, IL-1β, IFNγ et de TNFα [22]. Or, le parasite, arrêté dans les plexus choroïdes et l’organe circumventriculaire, ne pénètre pas le cerveau. Cette observation illustre l’existence de mécanismes de transfert de l’information, de l’extérieur vers l’intérieur du cerveau, de produire des cytokines. Au cours de l’infection avec le virus Borna, virus neurotrope de la maladie de Carré, des neurones participent à la production intracérébrale de TNF.
Le TGFβ, cytokine normalement absente du cerveau dont l’activité biologique dite anti-inflammatoire s’oppose à celle du TNFα et de l’IL-1, peut être exprimée lors de pathologies ischémiques, de traumatismes crâniens, d’encéphalites ou d’hyperactivité nerveuse excitatrice [23]. Elle serait produite par des cellules du cerveau lors de stress oxydatifs pour en limiter l’extension. A ce titre, les effets bénéfiques du TGFβ ont été démontrés sur des modèles expérimentaux de neurotoxicité et d’ischémie.
Au cours de la sclérose en plaque, l’IL-1 et le TNFα s’accumulent dans le LCR [24], particulièrement lors des poussées et dans les formes chroniques progressives. Par ailleurs, le TNFα est présent dans le cerveau, sur le site des plaques, principalement dans les astrocytes et les macrophages.
Modulation de différentes activités neuronales et mentales
Mémoire
L’IL-1 circulante peut agir sur les neurones de l’hippocampe en modulant la libération d’acétylcholine, neuromédiateur impliqué dans les processus de mémorisation. L’IL-1 inhibe les potentialisations à long terme (LTP), phénomène de sensibilisation synaptique associé aux mécanismes d’apprentissage et de mémorisation. Si la présence de l’IL-1β dans un cerveau normal est controversée, une synthèse faible mais constitutive d’IL-1β et d’IL-1Ra, fortement augmentée après une lésion ou une infection, semble bien admise (pour revue, voir [12]). D’autres protéines de la famille de l’IL-1, les IL-1 RacP sont également exprimées dans la plupart des régions du cerveau [25]. Une variante nouvelle récemment découverte dans cette famille moléculaire a pu être associée à une forme de retard mental liée à l’X. Son expression majeure dans l’hippocampe pendant la période de développement du cerveau suggère son rôle crucial dans le développement des fonctions cognitives [26]. L’activation de sa synthèse dans l’hippocampe sous l’effet de LTP souligne plus fortement encore cette implication dans le fonctionnement proprement neuronal [27]. Également actif sur la physiologie des neurones, le TNF peut moduler les courants calciques de certaines populations de neurones et l’efficacité de canaux potassiques [12].
Effets comportementaux
De nombreux travaux ont établi l’implication des cytokines dans la régulation du sommeil, en particulier au cours d’une infection. Les cytokines pro-inflammatoires (surtout IL-1β et TNFα) ont un rôle somnogénique [28] tandis que les cytokines anti-inflammatoires, IL-4 et IL-10 sont inhibitrices du sommeil. Des expériences de privation de sommeil effectuées sur des rats adultes montrent une augmentation de la production d’IL-1β dans des structures cérébrales bien définies: hypothalamus, hippocampe, cortex et mésencéphale.
Implication des cytokines dans certaines maladies mentales
Des travaux récents montrent que le stress prénatal induit des anomalies de structure persistantes de l’hippocampe et un déficit consécutif des performances d’apprentissage et de mémorisation [29]. Ces anomalies résultent d’une inhibition de la neurogenèse non seulement aux stades précoces de formation foetale mais tout au long de la vie. Une production massive d’hormones de stress par la mère pendant la période critique du développement serait responsable d’une réponse définitivement modifiée aux corticoïdes [30].
Des études réalisées sur des cohortes d’étudiants ont montré que les plus anxieux présentent une production de cytokines pro-inflammatoires (TNFα, IFNγ, IL-6) plus élevée et une production de cytokines anti-inflammatoires (IL-4, IL-10) plus faible. Un stress psychologique aigu, tel un examen universitaire, stimule la production de TNFα, d’IFNγ, d’IL-6, mais aussi d’IL-10. Des changements des concentrations des cytokines immunorégulatrices prendraient ainsi part aux réponses homéostatiques associées aux stress psychologiques [31].
De nombreuses études, tant cliniques qu’expérimentales, ont démontré dans la dépression une augmentation de la sécrétion de CRF et des cytokines circulantes, notamment des cytokines pro-inflammatoires IL-1β, IL-6 et IFNγ, [32], et une hyperactivité de l’axe HPA [33] avec hypercortisolémie. L’activation des cytokines pro-inflammatoires, en altérant la neurotransmission de la sérotonine, serait un des composants de la dépression. Toutefois, si le rôle du stress et l’implication des cytokines dans l’installation d’un état dépressif sont suggérés, la relation de cause à effet n’a pas été établie [34].
L’état de stress post-traumatique est caractérisé au niveau neurobiologique par une hyperactivité des systèmes sympathique et HPA, avec une hypersécrétion de CRF qui perdure et une hypocortisolémie. Bien que limitées, les recherches concernant les paramètres immunologiques montrent une augmentation de l’immunité humorale et cellulaire (revue dans [35]).
Plutôt qu’un processus d’auto-immunité, des caractéristiques spécifiques des cytokines IL-2 (et de leurs récepteurs) et IL-6 ont été corrélées à certaines formes de schizophrénie [36]. Leur relation avec une infection ou un traumatisme cérébral reste à déterminer.
La physiopathologie de la maladie d’Alzheimer fait intervenir à la fois des facteurs génétiques et d’environnement. Nous avons rappelé plus haut les effets toxiques de concentrations excessives en glucocorticoïdes sur les neurones de l’hippocampe, notamment ceux des aires CA1 et CA3. Or, ces neurones sont parmi les plus touchés dans la maladie d’Alzheimer et l’ischémie cérébrale. Le vieillissement est associé à une élévation des concentrations sériques de corticostérone qui pourrait, par l’effet d’une exposition chronique, accroître leur vulnérabilité aux neurotoxiques, au glutamate en particulier [9]. Par ailleurs, l’IL-1 est augmentée dans le cerveau de patients souffrant de la maladie d’Alzheimer [37]. L’activation microgliale par le peptide amyloïde en est le principal responsable, mais des concentrations élevées dans le sang peuvent être à l’origine d’une entrée de l’IL-1 ou du TNF dans le cerveau, soit du fait de lésions de la barrière hémato-encéphalique, soit par un passage actif à travers la barrière.
L’interprétation des données de la biologie dans les maladies mentales exige la plus grande prudence. Néanmoins, certaines observations donnent des pistes de recherche aidant à identifier les espaces communs aux grands systèmes. Ainsi, l’autisme, syndrome dont l’étiologie reste mal connue, a pu être associé dans certains cas à des colites ulcératives; une relation entre système nerveux et pathologie digestive inflammatoire a déjà été décrite dans la maladie de Parkinson. Des troubles obsessionnels compulsifs ont pu être associés à une auto-immunité post-infectieuse [38]. Des patients ayant reçu une injection contrôlée d’endotoxine développent des manifestations d’anxiété, des modifications de l’humeur et une baisse des capacités de mémorisation, parallèlement à une élévation des concentrations sériques de cytokines inflammatoires [39]. Ces travaux valident pour l’homme des observations déjà faites chez l’animal sur le modèle du comportement de maladie [40].
Connues jusqu’alors pour leurs rôles de messagers de l’immunité, les cytokines s’avèrent tenir dans le SNC des fonctions beaucoup plus diversifiées concernant la transmission nerveuse, la régénération neuronale et les équilibres ioniques. Le rôle du stress dans leur expression, et réciproquement leur fonction dans la modulation du processus d’adaptation, leurs relations directes avec les neuromédiateurs, voire leurs effets comportementaux les situent comme des acteurs centraux de l’interface entre stress, soma et psychisme. Dès lors, on doit regarder sous un jour nouveau les pathologies au cours desquelles la présence de cytokines dans le SNC ou le LCR a été observée. En effet, les cytokines d’origine myélo-monocytaires peuvent influer sur le fonctionnement des neurones et, réciproquement, la présence de cytokines dans le SNC au cours de certaines pathologies nerveuses pourrait être due à une synthèse neuronale et exercer des effets sur les cellules de l’immunité. Il est indispensable de prendre en compte cette potentielle diversité de cibles, et donc d’effets, des cytokines dans le SNC pour une compréhension plus exacte de ces maladies.
Appendices
Références
- 1. Sivadon P. Sur une théorie immunologique des maladies mentales. Ann Med Psychol 1964; 1: 369-82.
- 2. Delay J. Études de psychologie médicale. Paris: PUF, 1953.
- 3. Herbert T, Cohen S. Stress and immunity: a meta-analytic review. Psychosom Med 1993; 55: 364-79.
- 4. Francis D, Champagne F, Liu D, Meaney M. Maternal care, gene expression and the development of individual differences in stress reactivity. Ann NY Acad Sci 1999; 896: 66-84.
- 5. Kandel E. A new intellectual framework for psychiatry. Am J Psychiatry 1998; 155: 457-69.
- 6. Thurin JM. Une vie sans soi. Clinique et interprétation des maladies psychosomatiques. Paris: Frison-Roche, 1996 : 237 p.
- 7. Besedovsky H, Del Rey A. Immune-neuro-endocrine interactions: facts and hypotheses. Endocrinol Rev 1996; 17: 64-102.
- 8. Wooley C, Gould E, McEwen B. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res 1990; 531: 225-31.
- 9. Stein-Behrens B, Mattson MP, Chang I, Yeh M, Sapolsky R. Stress exacerbates neuron loss and cytoskeletal pathology in the hippocampus. J Neurosci 1994; 14: 5373-80.
- 10. Nadeau S, Rivier S. Regulation of the gene encoding TNFα in the rat brain and pituitary in response in different models of systemic immune challenge. J Neuropathol Exp Neurol 1999; 58: 61-77.
- 11. Holsboer F. Clinical Neuroendocrinology. In: Charney D, Nestler E, Bunney B, eds. Neurobiology of mental illness. Oxford : Oxford University Press, 1999 : 149-61.
- 12. Vitkovic L, Bockaert J, Jacque C. Inflammatory cytokines: neuromodulators in normal brain? J Neurochem 2000; 74: 457-71.
- 13. Tchélingérian J, Monge M, Le Saux F, Zalc B, Jacque C. Differential oligodendroglial expression of TNF receptors in vivo and in vitro. J Neurochem 1995; 65: 2377-80.
- 14. Benzing T, Brandes R, Sellin L, et al. Upregulation of RGS7 may contribute to TNF-induced changes in CNS function. Nat Med 1999; 8: 913-8.
- 15. Cartmell T, Luheshi G, Rothwell N. Brain sites of action of endogenous interleukin-1 in the febrile response to localized inflammation in the rat. J Physiol (Lond) 1999; 518: 585-94.
- 16. Dunn A, Wang J, Ando T. Effects of cytokines on cerebral neurotransmission. Comparison with the effects of stress. Adv Exp Med Biol 1999; 461: 117-27.
- 17. Marx C, Ehrhart-Bornstein M, Scherbaum W, Bornstein S. Regulation of adrenocortical function by cytokine-relevance for immune-endocrine interaction. Horm Metab Res 1998; 6-7: 416-20.
- 18. Loddick S, Turnbull A, Rothwell N. Cerebral IL-6 is neuroprotective during permanent focal cerebral ischemia in the rat. J Cereb Blood Flow Metab 1998; 18: 176-9.
- 19. Plata-Salaman C, Ilyin S, Turrin N, et al. Kindling modulates the IL-1β, TNFα, TGFβ-1 and neuropeptide mRNAs in specific brain regions. Brain Res Mol Brain Res 2000; 75: 2248-58.
- 20. Wesselingh S, Power C, Glass J, et al. Intracerebral cytokine messenger RNA expression in AIDS dementia. Ann Neurol 1993; 33: 576-82.
- 21. Goodman J, Robertson C, Grossman R, Narayan R. Elevation of TNF in head injury. J Neuroimmunol 1990; 30: 213-7.
- 22. Quan N, Mhlanga J, Whiteside M, McCoy A, Kristensson K, Herkenham M. Chronic overexpression of proinflammatory cytokines and histopathology in the brains of rats infected with Trypanosoma brucei. J Comp Neurol 1999; 414: 114-30.
- 23. Lehrmann E, Kiefer R, Finsen B, Diemer N, Zimmer J, Hartung H. Cytokines in cerebral ischemia: expression of TGFβ1 mRNA in the postischemic adult rat hippocampus. Exp Neurol 1995; 131: 114-23.
- 24. Hauser S, Doolittle T, Lincoln R, Brown R, Dinarello C. Cytokine accumulations in CSF of multiple sclerosis: frequent detection of IL-1 and TNF but not IL-6. Neurology 1990; 40: 1735-40.
- 25. Ilyin S, Gayle D, Flynn M, Plata-Salaman C. IL-1β system (ligand, receptor type I, receptor accessory protein and receptor antagonist), TNFα, TGFβ1 and neuropeptide Y mRNAs in specific brain regions during bacterial LPS-induced anorexia. Brain Res Bull 1998; 45: 507-15.
- 26. Carrié A, Jun L, Bienvenu T, et al. A new member of the IL-1 receptor family highly expressed in hippocampus and involved in X-linked mental retardation. Nat Genet 1999; 23 : 25-31.
- 27. Schneider H, Pitossi F, Balschun D, Wagner A, del Rey A, Besedovsky HO. A neuromodulatory role of interleukin-1beta in the hippocampus. Proc Natl Acad Sci USA 1998; 95: 7778-83.
- 28. Krueger J, Fang J, Taishi P, Chen Z, Kushikata T, Gardi J. Sleep. A physiologic role for IL-1β and TNF-α. Ann NY Acad Sci 1998; 856: 148-59.
- 29. Lemaire V, Koehl M, Le Moal M, Abrous D. Prenatal stress produces learning deficits associated with an inhibition of neurogenesis in the hippocampus. Proc Natl Acad Sci USA 2000; 97: 11032-37.
- 30. McEwen B, Magarinos AM. Stress and hippocampal plasticity: implications for the physiopathology of affective disorders. In: Hindmarch I, McEwen B, eds. Human psychopharmacology: clinical and experimental. New York : John Wiley and Sons Publishers, 2001 : S7-19.
- 31. Maes M, Song C, Lin A, et al. The effects of psychological stress on humans: increased production of pro-inflammatory cytokines and a Th1-like response in stress-induced anxiety. Cytokine 1998; 10: 313-8.
- 32. Maes M, Bosmans E, Meltzer H, Scharpé S, Suy E. Interleukin-1β: a putative mediator of HPA axis hyperactivity in major depression? Am J Psychiatry 1993; 150: 1189-93.
- 33. Connor T, Leonard B. Depression, stress and immunological activation: the role of cytokines in depressive disorders. Life Sci 1998; 62: 583-606.
- 34. De Beaurepaire R. Dépression et cytokines. Dépression 1999; 17 : 40-52.
- 35. Newport J, Nemeroff C. Neurobiology of posttraumatic stress disorder. Curr Opin Neurobiol 2000; 10: 211-8.
- 36. Griffin W, Stanley L, Ling C, et al. Brain IL-1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA 1989; 86: 7611-5.
- 37. Villemain F, Chatenoud L, Galinowski A, et al. Aberrant T-cell-mediated immunity in untreated schizophrenic patients: deficient interleukin-2 production. Am J Psychiatry 1989; 146: 609-16.
- 38. Cohen D, Flament M. Troubles obsessionnels compulsifs infanto-juvéniles et pathologies immunes. Perspectives Psy 2000; 39: 372-6.
- 39. Reichenberg A, Yirmiya R, Schuld A, et al. Cytokine-associated emotional and cognitive disturbances in humans. Arch Gen Psychiat 2001; 58: 445-52.
- 40. Dantzer R, Aubert A, Bluthé RM, et al. Sickness behaviour: a neuroimmune-based response to infectious disease. I. Neuro-immune interactions. In: Patterson P, Kordon C, Christen Y, eds. Neurologic and psychiatric disorders. Berlin Heidelberg: Springer-Verlag, 2000 : 169-84.
List of figures
Figure 1
Représentation schématique des interventions des cytokines dans le fonctionnement de l’axe du stress.
Les neurones parvocellulaires du noyau paraventriculaire de l’hypothalamus produisent la CRH, hormone également abondante dans l’amygdale. Cet autre site joue un rôle crucial dans la perception de peur et d’anxiété et la production de noradrénaline qui stimule les neurones du noyau paraventriculaire de l’hypothalamus. Dans des conditions normales, l’action de la CRH sur les cellules corticotrophiques de l’hypophyse induit et règle la sécrétion de l’ACTH, soit en l’augmentant à la suite d’un stress, soit en la réduisant en fonction de la concentration élevée de corticoïdes. Les effets de la CRH sont eux-mêmes modulés par la vasopressine et par certaines cytokines. Véhiculé par la circulation sanguine, l’ACTH agit sur les corticosurrénales qui, en réponse, produisent le cortisol. Ce dernier, utilisant également la voie sanguine, agit notamment sur l’hypothalamus, sur l’hippocampe et sur l’amygdale et participe à la régulation de l’immunité.
Figure 2
Représentation schématique des relations entre cytokines intra-cérébrales et pathologies nerveuses et/ou psychiques.
En regard de chaque groupe de pathologies figure la liste des cytokines impliquée dans une ou plusieurs de ces pathologies. Les lettres IC dans les ronds rouges indiquent que la présence des cytokines dans le cerveau a été démontrée. Le point d’interrogation indique une absence de démonstration de cette présence. Les références sont données dans le texte ou, à défaut, citées dans [12].