Abstracts
Résumé
Nous avons comparé certaines caractéristiques démographiques des familles de 221 sujets atteints de la maladie d’Alzheimer (MA) recrutés au Saguenay-Lac-Saint-Jean et celles de 218 témoins sélectionnés dans la même région et ne présentant aucune atteinte cognitive. Notre étude repose sur l’hypothèse de l’existence d’un lien entre la MA et certains aspects de l’histoire démographique des individus atteints. Les données démographiques proviennent du fichier de population BALSAC. L’analyse du rapport de masculinité à la naissance (RMN) met en lumière un excédent de filles dans les fratries des sujets atteints de MA. Étant donné qu’une incidence plus élevée de MA chez les femmes a été observée à de multiples reprises, nous avons postulé qu’un facteur familial pouvait opérer différemment selon le sexe et avoir un impact sur le RMN et sur le risque de développer la MA. Nous avons aussi observé que la mortalité infantile était significativement plus faible dans les fratries des individus atteints porteurs de l’allèle APOE-ε4. Ces observations laissent croire à la présence d’un facteur familial qui aurait un effet sur le risque de MA et sur la mortalité infantile.
Abstract
We performed an analysis of demographic factors among 221 autopsy-confirmed Alzheimer disease (AD) subjects recruited in the Saguenay region of Québec and among 218 controls selected in the same area and free of any cognitive impairment. We hypothesized that a relationship could exist between AD and the variables under study leading to differential demographic characteristics among affected and control families. Demographic information was obtained from the BALSAC population register. Analysis of sex ratio at birth indicated that AD cases’ sibships contained an excess of females. As a higher incidence of AD among women has repeatedly been reported, we postulate that a family factor could operate differentially according to gender and have an impact on sex ratio at birth through selective conception or foetus wastage and on the risk of developing AD. We also found that infant mortality was markedly lower among sibships of cases who were APOE-ε4 carriers. These observations suggest the presence of a family factor that would have an effect on the risk of AD and on infant mortality hazards.
Article body
La maladie d’Alzheimer (MA) se caractérise par une perte progressive de la mémoire et une détérioration générale de toutes les fonctions cognitives aboutissant finalement à la démence. Au cours des années 1990, d’importantes découvertes sur le rôle des facteurs génétiques dans l’étiologie de la MA ont été effectuées. Trois gènes ont été identifiés sur les chromosomes 21 (Goate et al., 1991), 14 (Sherrington et al., 1995) et 1 (Levy-Lahad et al., 1995), et des mutations pouvant expliquer le développement de la maladie chez environ 85 % des familles caractérisées par un âge d’apparition précoce ont été décrites (Rocchi et al., 2003). Une forte association entre l’allèle ε4 du gène de l’apolipoprotéine E (APOE) et le sous-type plus commun de la maladie, caractérisé par un âge d’apparition tardif, a aussi été démontrée (Strittmatter et al., 1993; Saunders et al., 1993). Il est admis que d’autres facteurs génétiques agissant conjointement ou non avec l’APOE-ε4 jouent également un rôle dans la MA (Bertram et Tanzi, 2001). Le rôle exact à attribuer aux facteurs génétiques reste à préciser, surtout en ce qui concerne les cas sporadiques de la maladie, et d’autres composantes biologiques de même que certaines expositions environnementales (Grant et al., 2002) et caractéristiques socioéconomiques (Karp et al. 2004) ne peuvent être éliminées comme facteurs étiologiques.
Différents facteurs de risque ont été explorés dans les études épidémiologiques sur la MA (van Duijn, 1996; Ritchie et Lovestone, 2002). Certains éléments sont relatifs à l’histoire démographique des patients et de leurs familles. L’âge parental à la naissance des cas et le rang de naissance ont attiré l’attention à cause d’un lien possible entre la MA et le syndrome de Down (SD) et parce qu’un âge maternel avancé représente le principal facteur de risque du SD (Farrer et al., 1991). La plupart des individus atteints du SD qui survivent jusqu’à l’âge de 35 ans développent des modifications neuropathologiques identiques à celles observées dans la MA (Holland et Oliver, 1995). Dans une nouvelle analyse des premières études de cas témoins, van Duijn et ses collaborateurs (1991) ont constaté une fréquence plus élevée du SD dans les familles des sujets atteints de MA. Un risque plus élevé de MA a aussi été observé chez les jeunes mères d’un enfant atteint du SD (Schupf et al., 1994). La possibilité d’une association entre l’âge parental à la naissance et la MA a été investiguée dans de nombreuses études (Farrer et al., 1991; Rocca et al., 1991; Clarnette et al., 1992; Prince et al., 1994; The Canadian Study of Health and Aging, 1994; Whalley et al., 1995; Bertram et al. 1998; Ptok et al. 2000). Cependant, aucune conclusion définitive ne peut être tirée car les résultats sont très différents d’une étude à l’autre.La fécondité et la mortalité chez les sujets atteints de MA et leurs apparentés au premier degré ont été analysées sous l’hypothèse qu’un facteur d’ordre génétique ou environnemental mis en cause dans l’étiologie de la maladie pourrait également avoir un impact sur certains aspects de la mortalité ou de la fécondité chez les membres de la famille exposés ou porteurs. Peu d’études ont exploré cette avenue et, là encore, les résultats ne permettent pas de conclure dans une direction précise (Heston et Mastri, 1977; Heston et al., 1981; Whalley et al., 1982; Corkin et al., 1983; White et al., 1986). Plus récemment, Ptok et ses collègues (2002), s’intéressant à la possibilité d’un effet protecteur de l’estrogène, ont comparé le nombre moyen d’enfants d’un groupe de sujets Alzheimer et d’un groupe témoin et n’ont trouvé aucune différence significative.
Le faible nombre d’études et le manque d’uniformité des résultats pourraient en partie s’expliquer par des difficultés à collecter des données fiables concernant les histoires démographiques familiales (Clarnette et al., 1992). La connaissance de ces histoires par les informateurs est variable car elle dépend, notamment, de leur relation avec les individus atteints; par conséquent, la collecte des données est sujette à un biais de mémoire et la qualité de l’information peut varier énormément parmi les cas et les témoins (Rocca et al., 1991). Dans la région du Saguenay-Lac-Saint-Jean (SLSJ), ce problème peut être surmonté grâce au fichier de population informatisé BALSAC, qui relie tous les registres paroissiaux concernant les baptêmes, les mariages et les sépultures qui ont eu lieu dans la région depuis son ouverture au peuplement d’origine européenne, en 1838. Le fichier BALSAC permet la reconstitution des familles et l’analyse de leur histoire démographique.
Une étude à caractère multidisciplinaire sur la MA a été effectuée au SLSJ par le groupe de recherche du Projet IMAGE (Gauvreau et al., 1988), et un registre contenant plus de 700 cas de MA a été constitué. Dans le cadre de notre étude, l’histoire familiale des événements démographiques a été reconstruite pour les 221 cas de MA dont le diagnostic avait pu être confirmé par autopsie. Nous nous sommes intéressés à l’âge des parents à la naissance et au rang de naissance, à la fécondité des sujets atteints et à celle de leurs parents, à la mortalité infantile parmi la fratrie et la descendance de ces sujets ainsi qu’à la mortalité chez leurs parents. Les résultats ont été comparés à ceux obtenus auprès de 218 témoins sélectionnés dans la même région et ne souffrant d’aucune atteinte cognitive. Nous avons formulé l’hypothèse qu’il existe entre la MA et les variables étudiées un lien conduisant à observer des caractéristiques démographiques différentes dans les familles des malades et dans celles des témoins.
Matériel et méthodes
Cas
Les procédures diagnostiques du Projet IMAGE étaient basées sur les critères NINCDS-ADRDA (McKhann et al., 1984) et sur des critères morphologiques et morphométriques publiés antérieurement (Khachaturian, 1985; Tiberghien et al., 1993). Nous avons réalisé notre étude auprès d’une cohorte de 221 cas de MA décédés entre 1987 et 1993 et dont le diagnostic a été confirmé à l’autopsie. Comme il est généralement admis que les études épidémiologiques sur les facteurs de risques mis en cause dans l’étiologie de la MA ont été peu concluantes jusqu’à maintenant, en partie à cause de l’hétérogénéité de la maladie, notre échantillon a été analysé dans son entier et a aussi été divisé en sous-groupes selon l’âge d’apparition de la maladie, le sexe et le génotype au locus de l’APOE. Notre objectif, en procédant ainsi, était d’augmenter nos chances de travailler sur des groupes plus homogènes, en faisant l’hypothèse que les variables choisies correspondent à des facteurs étiologiques spécifiques. Pour effectuer les analyses par sous-groupes basés sur l’âge d’apparition, nous avons adopté le point de coupure couramment utilisé (Rocchi et al., 2003) : les cas ont été classés comme précoces si la maladie était apparue avant 65 ans et tardifs si elle s’était manifestée à 65 ans ou après. Les génotypes au locus de l’APOE ont été regroupés selon la présence ou l’absence de l’allèle ε4, celui qui confère une susceptibilité à la MA. Notre objectif était de prendre en considération la possibilité que d’autres facteurs de risque puissent opérer conjointement avec l’allèle ε4 du gène APOE ou en son absence.
Témoins
Une étude sur la santé et le vieillissement a été entreprise en 1994 dans la région du SLSJ. Elle a notamment permis de définir un groupe témoin composé d’individus âgés de plus de 70 ans au moment de l’étude et ne souffrant d’aucune atteinte cognitive selon une évaluation neuro-psychologique (Gauthier et al., 2000). Nous avons choisi nos témoins dans ce groupe en les appariant aux cas selon le sexe et l’année de naissance, à l’intérieur d’un intervalle de 5 ans. En utilisant ces critères, nous avons pu sélectionner 218 témoins. Les caractéristiques des cas et des témoins concernant le sexe, le lieu et l’année de naissance, les fréquences alléliques au locus de l’APOE et, pour les cas, l’âge à l’apparition de la maladie et au décès, apparaissent au tableau 1. Les cas d’âge d’apparition précoce et d’âge d’apparition tardif sont décrits séparément. Les fréquences des trois allèles du gène APOE présentent des différences significatives chez les cas et chez les témoins (χ2 = 51,5; d. l. 2; p < 0,00001). Comme on s’y attendait, la fréquence de l’allèle ε4 est plus élevée chez les cas (0,40) que chez les témoins (0,10); elle l’est à la fois chez les cas d’âge d’apparition précoce et chez les cas d’âge d’apparition tardif (0,42 et 0,40 respectivement). Les fréquences ont été calculées d’après les génotypes de 190 cas et de 90 témoins.
Tableau 1
Description de l’échantillon

Moyenne ± écart type (étendue)
Les allèles correspondent aux différentes variantes d’un gène.
*** : pas d’âge aux premiers symptômes et au décès.
Données démographiques
Nous avons mentionné que le fichier de population BALSAC contient les enregistrements informatisés des actes de baptême, mariage et sépulture rédigés au Saguenay, depuis son ouverture au peuplement d’origine européenne, en 1838, jusqu’en 1971; ce registre contient aussi certains décès jusqu’en 1986. Les cas et les témoins ont été localisés dans le fichier BALSAC à l’aide de leur date de naissance ou de leur date de mariage et du nom de leur conjoint(e), ou encore à l’aide des noms et de la date de mariage de leurs parents. Nous avons pu retracer 216 cas ainsi que 193 témoins. Deux séries de données ont ainsi été extraites du fichier de population. La première série contient de l’information sur les sujets et leur famille d’origine, y compris leurs parents et leurs frères et soeurs. Nous avons obtenu pour chaque sujet, quand elles étaient disponibles, les dates de naissance, mariage et décès de ses parents, de même que les dates de naissance et de décès et le sexe de chacun de ses frères et soeurs. Nous avons vérifié l’apparentement au premier degré chez les cas et avons découvert dans notre échantillon trois paires de frères ou soeurs, une paire de demi-frères et un groupe de trois soeurs. Nous avons conservé aux fins d’analyse un seul cas par fratrie afin de contrôler tout biais de recrutement et d’éviter que certaines familles soient surreprésentées. Nous avons sélectionné au hasard un membre dans chacune de ces familles, ce qui nous a finalement donné un échantillon composé de 210 cas. Nous avons aussi trouvé un couple de frère et soeur parmi les témoins et un seul de ces individus a été conservé. Par ailleurs, nous avons pu établir que deux témoins étaient les frères ou soeurs de deux sujets atteints et nous les avons exclus de l’étude.
La deuxième série de données contient de l’information sur la propre famille des sujets. Cette fois, nous avons travaillé avec tous les sujets sans prendre en considération leur apparentement au premier degré, étant donné que chaque sujet forme une famille distincte avec son conjoint ou sa conjointe et ses enfants. Pour chaque sujet nous avons obtenu, lorsque c’était possible, ses dates de naissance et de mariage, la date de décès de son conjoint ou de sa conjointe, et les dates de naissance et de décès ainsi que le sexe de chacun de ses enfants.
Parmi les 216 cas et 193 témoins retracés dans le fichier de population BALSAC, et en conservant un seul sujet par fratrie dans les familles d’origine, nous avons obtenu des données sur l’histoire démographique de 195 cas et 166 témoins concernant leur propre famille, et de 193 cas et 176 témoins relativement à leur famille d’origine. Pour chaque paramètre analysé, la taille de l’échantillon a donc varié selon le nombre de sujets pour lesquels les données nécessaires aux calculs étaient disponibles. Puisque notre objectif était de comparer les cas et les témoins, nous n’avons aucune raison de croire que la quantité et la qualité de l’information disponible différaient entre ces deux groupes et que nos résultats peuvent être biaisés.
Tableau 2
Nombre et dates des événements démographiques retracés dans le fichier BALSAC chez les cas et chez les témoins

Enfin, même si toutes les histoires démographiques familiales sont traitées comme si l’ensemble des familles formait une seule et unique cohorte, ces événements s’étendent en fait sur une longue période de temps; cependant, nous avons comparé les périodes pour les cas et les témoins et, comme le montre le tableau 2, elles sont très semblables pour les deux groupes. Par conséquent, toute différence observée entre les deux groupes ne devrait pas avoir été introduite par des événements démographiques survenus dans des contextes temporels différents.
Analyse de l’âge parental et du rang de naissance
Nous avons obtenu l’âge parental à la naissance en calculant la différence entre la date de naissance de chaque sujet et les dates de naissance de ses père et mère. Le rang de naissance des sujets a été établi dans les familles dont l’histoire démographique a été estimée complète, c’est-à-dire lorsque la date de mariage, la date de décès d’au moins un des conjoints et les dates de naissance de tous les enfants présents dans le fichier BALSAC étaient connues. Le calcul du rang de naissance d’un sujet tient uniquement compte de ses frères et soeurs germains. Nous avons comparé les résultats avec un test t de Student bilatéral pour des échantillons indépendants. Toutes les analyses statistiques ont été effectuées avec le logiciel SPSS. Pour chaque variable, nous avons d’abord comparé l’ensemble des cas et des témoins, puis nous avons effectué la comparaison des cas d’âge d’apparition précoce et des cas d’âge d’apparition tardif séparément avec les témoins. Les cas et témoins de même sexe ont aussi été comparés séparément. Enfin, les cas porteurs de l’allèle APOE-ε4 ont été comparés aux témoins qui étaient aussi porteurs et les cas non porteurs ont été comparés aux témoins qui n’étaient pas porteurs de l’allèle.
Analyse de la fécondité
Pour les familles d’origine des sujets, nous avons effectué l’analyse de la fécondité dans les familles complètes, c’est-à-dire celles pour lesquelles nous connaissions la date de mariage et la date de décès d’au moins un des conjoints. Seuls les frères et soeurs germains des sujets ont été pris en considération (les demi-frères et demi-soeurs n’ont pas été retenus). Pour l’étude des enfants des sujets, nous avons considéré toutes les familles ayant une date de mariage inscrite dans le fichier. Comme tous les sujets de l’étude ont été recrutés après la fin de leur vie reproductive, nous avons fait l’hypothèse que si une date de mariage était présente dans le fichier BALSAC, le fichier contenait l’histoire complète de la vie reproductive de la famille. Le calcul de la taille de la famille a été basé sur tous les enfants présents dans les documents informatisés sans égard au fait que leur identification ait été établie par l’enregistrement de leur naissance (baptême) ou d’un autre événement démographique. Nous avons calculé le rapport de masculinité en faisant le rapport des naissances masculines sur les naissances féminines; dans les familles d’origine, les calculs ont été basés sur les frères et soeurs, excluant les sujets eux-mêmes de même que quelques naissances de sexe indéterminé. La comparaison statistique de la taille moyenne des familles a été effectuée à l’aide d’un test t de Student bilatéral pour échantillons indépendants. Nous avons aussi comparé la proportion de naissances de chaque sexe par rapport au nombre de naissances total en utilisant le test du χ2 de Pearson avec un degré de liberté.
Analyse de la mortalité infantile
Nous avons défini la mortalité infantile comme étant tous les décès survenus avant le premier anniversaire. Parmi la fratrie des sujets, nous avons considéré un enfant seulement si sa date de naissance était connue et si le décès d’un de ses parents était présent dans le fichier BALSAC tout en ayant eu lieu au moins un an après la naissance de cet enfant. Notre but était de prendre en considération seulement les enfants encore présents dans la région un an après leur naissance et dont le décès aurait par conséquent été enregistré s’il avait eu lieu. Nous avons procédé de cette manière afin d’éviter le plus possible de sous-estimer la mortalité. Dans l’étude de la mortalité infantile chez les enfants des sujets, nous avons jugé qu’étant donné que tous les sujets avaient été recrutés après leurs années de vie reproductive, le décès d’un enfant durant sa première année de vie aurait été indiqué dans le fichier BALSAC. Les taux de mortalité ont été estimés pour l’ensemble des naissances et séparément pour les naissances masculines et féminines. Nous avons utilisé le test du χ2 de Pearson avec un degré de liberté pour comparer les groupes.
Analyse de la mortalité adulte
Nous avons étudié la mortalité dans les familles des sujets en utilisant l’âge parental au décès. Celui-ci a été calculé au moyen des dates de naissance et de décès des parents des sujets, ce qui veut dire que l’âge parental au décès n’était disponible que pour les parents nés et décédés au SLSJ. Nous avons comparé statistiquement les résultats entre cas et témoins avec un test t de Student bilatéral pour échantillons indépendants.
Résultats
Analyse de l’âge parental et du rang de naissance
Les résultats concernant l’âge parental à la naissance et le rang de naissance des sujets apparaissent au tableau 3. L’âge maternel et l’âge paternel au décès sont légèrement plus élevés chez les cas que chez les témoins, et le rang de naissance est légèrement plus faible. Nous n’avons constaté que de très faibles variations entre les sous-groupes et aucune différence significative au seuil de 0,05.
Tableau 3
Âge parental et rang de naissance chez les cas et les témoins

Analyse de la fécondité et du rapport de masculinité
Les résultats apparaissent au tableau 4 pour les familles d’origine des sujets : la taille moyenne de la famille est identique chez les cas et les témoins. On retrouve en moyenne, dans les familles d’origine des cas précoces, un enfant de moins que dans celles des cas tardifs et des témoins. Cette différence n’est cependant pas significative. Dans tous les autres sous-groupes, nous n’observons que de légères variations et n’avons trouvé aucune différence statistiquement significative.
L’étude du rapport de masculinité chez les frères et soeurs des sujets fait apparaître des différences intéressantes. Les fratries des cas Alzheimer contiennent significativement plus de filles que les fratries des témoins; de plus, lorsque nous subdivisons les cas selon l’âge d’apparition de la maladie, nous découvrons que cet excédent de naissances féminines caractérise principalement les cas précoces. Cette différence entre cas et témoins est significative pour le groupe entier (p = 0,02) et pour les cas précoces (p = 0,008). La subdivision des cas et des témoins selon le sexe et selon la présence ou l’absence de l’allèle APOE-ε4 montre que cette différence est plus marquée et devient statistiquement significative chez les sujets de sexe féminin (p = 0,007) et chez les sujets et les témoins qui ne possèdent pas l’allèle APOE-ε4 (p = 0,05).
Tableau 4
Fécondité dans les familles d’origine des cas et des témoins

0,005 < p ≤ 0,05.
Le tableau 5 présente les résultats des analyses de la fécondité des cas et des témoins. Le nombre moyen d’enfants est plus élevé chez les premiers et cette différence devient significative quand on compare les cas tardifs et les témoins (p = 0,04); en effet, les cas tardifs ont en moyenne un enfant de plus que les témoins. Le regroupement des cas et des témoins selon le sexe et selon la présence ou l’absence de l’allèle APOE-ε4 met en évidence de faibles variations entre les groupes, mais on n’observe aucune autre différence statistiquement significative.
Table 5
Fécondité des cas et des témoins

0,005 < p ≤ 0,05.
Tableau 6
Taux (‰) de mortalité infantile dans les fratries des cas et des témoins
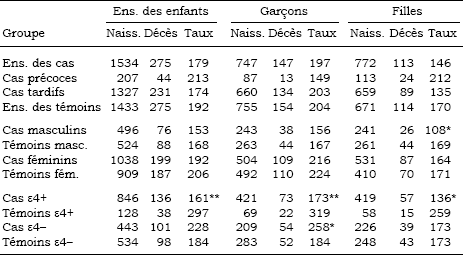
Note : * 0,005 < p ≤ 0,05, ** p ≤ 0,005.
L’étude du rapport de masculinité chez les enfants des sujets indique que les familles des cas contiennent plus de garçons, et celles des témoins plus de filles. Cette différence est statistiquement significative quand on compare les cas tardifs et les témoins (p = 0,04). Les analyses des regroupements selon le sexe et l’allèle APOE-ε4 ne révèlent aucune différence significative entre cas et témoins.
Analyse de la mortalité infantile
On trouvera les taux de mortalité infantile au tableau 6 pour les frères et soeurs des cas et des témoins, et au tableau 7 pour leurs enfants. Les résultats sont présentés pour toutes les naissances et pour les naissances féminines et masculines séparément. La somme des naissances masculines et féminines n’est pas égale au total des naissances parce que, pour un petit nombre de naissances (et de décès), le sexe de l’enfant était indéterminé. Dans la fratrie des sujets, la mortalité infantile est légèrement plus faible chez les cas que chez les témoins. Le regroupement selon l’âge d’apparition de la maladie et le sexe n’introduit pas de différences notables, sauf en ce qui concerne les sujets de sexe masculin, pour qui la mortalité infantile des soeurs est significativement plus faible chez les cas que chez les témoins (p = 0,05). Une différence importante apparaît quand nous comparons les cas et les témoins porteurs d’un allèle APOE-ε4: la mortalité infantile y est significativement plus faible chez les frères et soeurs des cas que chez les frères et soeurs des témoins (p = 0,0002); cette différence demeure significative quand les naissances sont regroupées selon le sexe mais elle est un peu plus marquée chez les garçons (p = 0,005 pour les garçons et p = 0,01 pour les filles). À l’opposé, chez les sujets non porteurs de l’allèle APOE-ε4, la mortalité infantile est plus élevée dans la fratrie des cas que dans celle des témoins et la différence est significative pour les garçons (p = 0,05).
Tableau 7
Taux (‰) de mortalité infantile chez les enfants des cas et des témoins

0,005 < p ≤ 0,05.
La mortalité infantile est un peu plus faible chez les enfants des cas que chez ceux des témoins. Quand nous regroupons les cas selon l’âge d’apparition de la maladie, nous observons que cette différence est due aux familles des cas précoces, et spécialement aux naissances féminines, où le taux de mortalité est significativement plus faible chez les enfants des cas que chez ceux des témoins (p = 0,02). Quand les sujets sont regroupés selon le sexe, nous constatons que, chez les sujets de sexe masculin, la mortalité infantile est plus faible chez les cas que chez les témoins, et cette différence devient significative pour les naissances de garçons (p = 0,03); chez les sujets de sexe féminin, la mortalité est légèrement plus élevée dans les familles des cas que dans celles des témoins. Le regroupement des cas selon la présence ou l’absence de l’allèle APOE-ε4 ne fait apparaître aucune différence notable.
Table 8
Âge au décès des parents des cas et des témoins

Analyse de la mortalité adulte
Le tableau 8 présente les résultats obtenus dans l’étude de l’âge parental au décès. L’âge moyen au décès est inférieur d’environ 3 ans chez les pères des cas comparativement aux pères des témoins, et de 2 ans chez les mères, mais ces différences ne sont pas significatives à un niveau de 0,05. Cet âge inférieur au décès chez les parents des cas est plus marqué quand on considère l’âge d’apparition de la maladie : les pères des cas précoces meurent en moyenne 5 ans plus tôt que les pères des témoins, et la différence atteint 7 ans chez les mères. Ces différences ne sont cependant pas significatives. Aucune des différences observées dans les autres regroupements n’atteint le seuil de signification statistique de 0,05.
Discussion
Cette étude avait pour but de vérifier les associations possibles entre des facteurs démographiques et la MA. Notre recherche présente plusieurs points forts : le diagnostic de MA est certain car il a été confirmé par autopsie chez tous les cas, les témoins ont été sélectionnés dans la population et ne présentaient aucun déficit cognitif au moment de l’étude; les données démographiques proviennent d’une fichier de population validé qui s’appuie sur des registres paroissiaux. Sa principale faiblesse concerne la taille échantillonnale. En effet, même si elle portait initialement sur une série de cas relativement grande (n = 221), deux facteurs ont contribué à réduire la taille de l’échantillon. D’abord, pour chaque facteur étudié, nous ne pouvions inclure dans les analyses que les sujets pour lesquels les données nécessaires aux calculs étaient disponibles dans le fichier BALSAC. Cela signifie que nous avons pu étudier uniquement les sujets ayant vécu les événements démographiques concernés dans la région du SLSJ. Pour calculer l’âge du père à la naissance, par exemple, nous avions besoin des dates de naissance du sujet et de son père; si une de ces deux dates était manquante, nous devions exclure le sujet de nos analyses. Deuxièmement, afin d’examiner la possibilité qu’un facteur de risque potentiel affecte uniquement un sous-ensemble de cas, nous avons effectué les analyses sur des sous-groupes qui, à quelques occasions, étaient de taille assez réduite. Par le fait même, des différences intéressantes ont pu ne pas être détectées à cause d’un manque de puissance statistique. D’un autre côté, les niveaux de signification devraient être interprétés avec prudence puisqu’ils n’ont pas été ajustés pour le nombre de tests effectués.
Âge parental et rang de naissance
Nos données ne font ressortir aucune association entre l’âge du père ou de la mère à la naissance et la MA. De plus, nous n’avons observé aucun lien entre le rang de naissance et la MA. Le regroupement des sujets selon l’âge d’apparition de la maladie, le sexe et la présence ou l’absence de l’allèle APOE-ε4 n’a pas révélé de différence notable entre les cas et les témoins.
Dans la littérature, la relation entre l’âge parental à la naissance et la MA demeure controversée. Des résultats contradictoires ressortent des données publiées jusqu’à 1990 puisque quatre études constatent une association positive entre un âge maternel avancé et la MA, alors que huit autres ne font ressortir aucun lien (Clarnette et al., 1992). Dans une méta-analyse de données provenant d’études antérieures, Rocca et son équipe (1991) ont observé qu’un âge maternel avancé et un jeune âge maternel pouvaient constituer des facteurs de risque pour la MA. Trois études signalent un effet de l’âge paternel sur la MA : selon Farrer et ses coauteurs (1991), un jeune âge paternel pourrait être mis en cause dans l’étiologie des cas tardifs, via un mécanisme d’empreinte génétique, alors que Whalley et ses collègues (1995) ont postulé que des mutations spontanées se produisant chez les pères plus âgés pourraient avoir un effet sur les cas précoces de sexe masculin. Bertram et ses collègues (1998) ont aussi observé un âge paternel plus élevé dans un sous-groupe de sujets ayant une faible probabilité d’être porteurs d’un gène majeur de susceptibilité à la MA. Enfin, quatre autres études n’ont découvert aucun effet de l’âge parental sur le risque de développer la MA (Clarnette et al., 1992; Prince et al., 1994; The Canadian Study of Health and Aging, 1994; Ptok et al., 2000).
Notre étude ne confirme pas l’hypothèse voulant que l’âge parental et le rang de naissance soient des facteurs de risque pour la MA. Comme nous l’avons signalé précédemment, une taille échantillonnale réduite, particulièrement dans les analyses par sous-groupes, a pu nous empêcher de détecter certaines différences entre les cas et les témoins. Il se peut aussi que, comme dans l’étude de Bertram et al. (1998), l’effet de l’âge parental ne concerne qu’une partie des sujets et que notre choix de critères pour faire le regroupement des cas n’ait pas été approprié. Ces facteurs pourraient aussi expliquer les résultats contradictoires obtenus dans les autres études : en effet, si l’âge parental à la naissance joue un rôle uniquement dans un sous-groupe de cas, il est possible que, pour des raisons liées au plan d’étude ou à l’étiologie de la maladie, l’effet soit détectable dans certaines études mais non dans d’autres.
Fécondité et rapport de masculinité
Le nombre moyen d’enfants ne varie pas de façon importante chez les cas et les témoins. La seule différence significative observée concerne les cas tardifs, qui ont en moyenne un enfant de plus que les témoins. Nous avons retracé quatre études évaluant la fécondité dans les familles où la MA était présente. Heston et al. (1981) ainsi que Ptok et al. (2002) n’ont constaté aucune différence dans le nombre d’enfants de sujets et de leurs frères et soeurs. Les données de Whalley et de ses coauteurs (1982) ont montré que la taille moyenne de la famille des femmes chez qui la MA était apparue à un âge précoce représentait 76 % de celle des témoins, mais ce résultat n’était pas statistiquement significatif. Finalement, White et ses collègues (1986) ont montré dans leur étude que les hommes atteints de MA avaient significativement plus d’enfants que les témoins. Nous croyons que ces résultats prêchent en faveur d’un plus grand nombre d’études basées sur des tailles échantillonnales plus importantes.
Notre analyse du rapport de masculinité a révélé des résultats intéressants. Les fratries des cas contiennent un excès de filles quand on les compare aux fratries des témoins. Cette variation demeure significative lorsque les cas précoces, les cas de sexe féminin et les cas non porteurs de l’allèle APOE-ε4 sont analysés séparément. Inversement, chez les enfants des cas, nous trouvons en moyenne plus de garçons que chez les enfants des témoins. Cette différence est particulièrement évidente dans les familles des cas tardifs. À notre connaissance, notre étude est la première à explorer le rapport de masculinité à la naissance dans les familles où on retrouve des cas de MA. Cependant, l’incidence différente de la MA selon le sexe est une question débattue depuis longtemps (Ruitenberg et al., 2001; Ritchie et Lovestone, 2002). De nombreuses études font état d’un excès de femmes parmi les malades apparentés au premier degré à un cas de MA (Farrer et al., 1995; Lautenschlager et al., 1996; Payami et al., 1996a, 1996b; Rao et al., 1996); ces preuves sont en faveur de l’existence de facteurs de risque de la MA spécifiques au sexe agissant soit indépendamment (Lautenschlager et al., 1996; Payami et al., 1996a; Rao et al., 1996), soit de concert avec le génotype APOE (Farrer et al., 1995; Payami et al., 1996b; Bretsky et al., 1999). Nous postulons que si un facteur familial peut influencer le risque de développer la MA selon le sexe, il peut aussi avoir un impact sur le rapport de masculinité à la naissance dans les familles affectées, par le biais d’une conception ou d’une mortalité intra-utérine sélectives.
Mortalité infantile
Nous avons observé que la mortalité infantile était légèrement plus faible dans les familles des cas de MA, autant chez leurs enfants que dans leur fratrie. Ce résultat contredit deux études précédentes où on avait démontré un excédent de mortalité infantile chez les apparentés au premier degré de patients atteints de MA (Heston et Mastri, 1977; Heston et al., 1981). Dans la présente étude, la mortalité infantile était sensiblement inférieure dans les fratries des cas porteurs de l’APOE-ε4. Cette observation suggère la présence d’un facteur familial relié à l’allèle APOE-ε4 pouvant avoir un effet à la fois sur le risque de MA et sur la mortalité infantile. Dans une étude antérieure portant sur la même cohorte, nous avons observé des niveaux d’apparentement et de consanguinité plus élevés chez les cas tardifs porteurs de l’allèle APOE-ε4 que chez les témoins, ce qui indique que ces individus ont une probabilité plus élevée d’avoir des gènes en commun (Vézina et al., 1999).
De nombreuses études ont été effectuées sur le rôle de l’apolipoprotéine E dans diverses pathologies (Messier, 2003; Puglielli et al., 2003) et sur ses effets sur l’espérance de vie aux âges avancés (Ewbank, 2004), mais à notre connaissance il n’existe aucune étude sur la possibilité d’un lien entre la mortalité infantile et l’APOE. Nous émettons l’hypothèse que, dans les familles des cas porteurs de l’APOE-ε4, les enfants pourraient être plus résistants aux agents infectieux durant leurs premières années de vie mais plus vulnérables aux pathologies associées au gène de l’APOE à un âge plus avancé. Cette différence n’a pas été observée chez les enfants des cas porteurs de l’APOE-ε4. Cependant, ces enfants sont nés en moyenne quelques décennies plus tard (voir le tableau 2), alors que les taux de mortalité infantile avaient considérablement diminué; cela pourrait avoir fait en sorte que l’effet protecteur de ce facteur passe inaperçu.
Mortalité adulte
L’âge au décès est moins élevé chez les parents des cas, particulièrement chez les parents des cas précoces, bien qu’aucune des différences entre cas et témoins ne soit statistiquement significative. Corkin et ses coauteurs (1983) n’ont constaté aucune différence significative de l’âge parental au décès entre cas et témoins, mais leur étude était basée sur 37 cas et 34 témoins. Nous avons étudié la mortalité adulte au moyen d’une analyse de l’âge moyen au décès des parents de sujets atteints de MA. Toutefois, la moyenne est influencée par les valeurs extrêmes; dans notre étude, certains parents sont décédés à un âge relativement jeune, probablement de causes accidentelles ou reliées à la maternité pour les femmes. La construction de tables de mortalité et l’utilisation de paramètres issus de ces tables (probabilités de survie, espérance de vie) constituent une meilleure approche pour l’étude de la mortalité. Un échantillon de taille supérieure serait nécessaire à l’examen de cette question et à une analyse plus détaillée de la mortalité adulte dans les familles atteintes de MA.
Conclusion
En résumé, nos données ne confirment pas les observations précédentes concernant une association entre l’âge parental à la naissance et la MA; elles mettent plutôt en évidence l’absence d’effet ou un effet qui concernerait seulement un petit groupe de malades que nous ne sommes pas parvenus à caractériser.
Notre analyse de la fécondité et de la mortalité dans les familles des sujets a produit des résultats intéressants sur le rapport de masculinité à la naissance et sur la mortalité infantile dans les fratries des cas de MA. Puisque ces résultats n’ont été signalés dans aucune étude antérieure, ils devraient être considérés comme préliminaires, mais nous croyons qu’ils soulèvent certaines questions qui mériteraient d’être abordées dans d’autres études.
Appendices
Remerciements
L’auteure remercie les participants et leurs familles, qui ont permis la réalisation de cette étude, ainsi que les membres du Projet IMAGE. La plupart des analyses contenues dans cet article ont été exécutées durant le doctorat de l’auteure, qui remercie aussi Hubert Charbonneau, son directeur, ainsi que Denis Gauvreau et Évelyne Heyer, ses codirecteurs. Ce travail a pu être effectué en partie grâce à une bourse doctorale du CRSH. Enfin, la participation de Lise Gobeil à la mise à jour de l’article et les commentaires constructifs reçus des lecteurs anonymes ont été appréciés.
Références bibliographiques
- BERTRAM, L., et R. E. TANZI. 2001. « Dancing in the dark? The status of late-onset Alzheimer’s disease genetics », Journal of Molecular Neuroscience, 17 : 127-136.
- BERTRAM, L., R. BUSCH, M. SPIEGL, N. T. LAUTENSCHLAGER, U. MULLER et A. KURZ. 1998. « Paternal age as a risk factor for Alzheimer disease in the absence of a major gene », Neurogenetics, 1 : 227-280.
- BTRESKY, P. M., J. G. BUCKWALTER, T. E. SEEMAN, C. A. MILLER, J. POIRIER, G. H. SCHELLENBERG, C. E. FINCH et V. W. HENDERSON. 1999. « Evidence for an interaction between apolipoprotein E genotype, gender, and Alzheimer disease », Alzheimer Disease and Associated Disorders, 13 : 216-221.
- CLARNETTE, R. M., D. W. MOLLOY, W. E. MCILROY, J. LEVER et L. REES. 1992. « Maternal age and Alzheimer’s disease », Dementia, 3 : 32-37.
- CORKIN, S., J. H. GROWDON et S. L. RASMUSSEN. 1983. « Parental age as a risk factor in Alzheimer’s disease », Annals of Neurology, 13 : 674-676.
- EWBANK, D. C. 2004. « The APOE gene and differences in life expectancy in Europe », The Journals of Gerontology. Series A, Biological Sciences and Medical Sciences, 59 : 16-20.
- FARRER, L. A., L. A. CUPPLES, L. CONNOR, P. A. WOLF et J. H. GROWDON. 1991. « Association of decreased paternal age and late-onset Alzheimer’s disease—An example of genetic imprinting? », Archives of Neurology, 48 : 599-604.
- FARRER, L. A., L. A. CUPPLES, C. M. VAN DUIJN, A. KURZ, R. ZIMMER, U. MÜLLER, R. C. GREEN, V. CLARKE, J. SHOFFNER, D. C. WALLACE, H. CHUI, S. D. FLANAGAN, R. DUARA, P. ST. GEORGE-HYSLOP, S. A. AUERBACH, L. VOLICER, J. M. WELLS, C. VAN BROECKHOVEN, J. H. GROWDON et J. L. HAINES. 1995. « Apolipoprotein E genotype in patients with Alzheimer’s disease: Implications for the risk of dementia among relatives », Annals of Neurology, 38 : 797-808.
- GAUTHIER, E., I. FORTIER, F. COURCHESNE, P. PEPIN, J. MORTIMER et D. GAUVREAU. 2000. « Aluminum forms in drinking water and risk of Alzheimer’s disease », Environmental Research, 84 : 234-246.
- GAUVREAU, D., R. BOUCHARD, S. GAUTHIER, J. MATHIEU, C. BOILY, A. CHOLETTE, Y. ROBITAILLE, P. BOUCHARD, N. BOUCHARD, L.-P. DOYON, M. GAUDREAULT, A. OUELLET, M. DUMONT, P. KISHKA, C. FOURNIER, J. NALBANTOGLU, G. LACOSTE-ROYAL, D. GAUTRIN, S. FRODA, M. DE BRAEKELEER, G. BOUCHARD et J. MORTIMER. 1988. « The IMAGE Project: A geographical laboratory for the investigation of multidisciplinary data », dans P. M. SINET, Y. LAMOUR et Y. CHRISTEN, éd. Research and Perspective in Alzheimer’s Disease. Heidelberg, Springer-Verlag : 40-50.
- GOATE, A., M.-C. CHARTIER-HARLIN, M. MULLAN, J. BROWN, F. CRAWFORD, L. FIDANI, L. GIUFFRA, A. HAYNES, N. IRVING, L. JAMES, R. MANT, P. NEWTON, K. ROOKE, P. ROQUES, C. TALBOT, M. PERICAK-VANCE, A. ROSES, R. WILLIAMSON, M. ROSSOR, M. OWEN et J. HARDY 1991. « Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease », Nature, 349 : 704-706.
- GRANT, W. B., A. CAMPBELL, R. F. ITZHAKI et J. SAVORY 2002. « The significance of environmental factors in the etiology of Alzheimer’s disease », Journal of Alzheimer’s disease, 4 : 179-189
- HESTON, L. L., et A. R. MASTRI. 1977. « The genetics of Alzheimer’s disease. Associations with hematologic malignancy and Down’s syndrome », Archives of General Psychiatry, 34 : 976‑981.
- HESTON, L. L., A. R. MASTRI, E. ANDERSON et J. WHITE. 1981. « Dementia of the Alzheimer type. Clinical genetics, natural history, and associated conditions », Archives of General Psychiatry, 38 : 1085-1090.
- HOLLAND, A. J., et C. OLIVER 1995. « Down’s syndrome and the links with Alzheimer’s disease », Journal of Neurology, Neurosurgery and Psychiatry, 59 : 111-114.
- KARP, A., I. KAREHOLT, C. QIU, T. BELLANDER, B. WINBLAD et L. FRATIGLIONI. 2004. « Relation of education and occupation-based socioeconomic status to incident Alzheimer’s disease », American Journal of Epidemiology, 159 :175-183.
- KHACHATURIAN, Z. S. 1985. « Diagnosis of Alzheimer’s disease », Archives of Neurology, 42: 1097-1105.
- LAUTENSCHLAGER, N. T., L. A. CUPPLES, V. S. RAO, S. A. AUERBACH, R. BECKER, J. BURKE, H. CHUI, R. DUARA, E. J. FOLEY, S. L. GLATT, R. C. GREEN, R. JONES, H. KARLINSKY, W. A. KUKULL, A. KURZ, E. B. LARSON, K. MARTELLI, A. D. SADOVNICK, L. VOLICER, S. C. WARING, J. H. GROWDON et L. A. FARRER. 1996. « Risk of dementia among relatives of Alzheimer’s disease patients in the MIRAGE study: What is in store for the oldest old? », Neurology, 46 : 641-650.
- LEVY-LAHAD, E., W. WASCO, J. P. POORKA, D. M. ROMANO, J. OSHIMA, W. H. PETTINGELL, C. YU, P. D. JONDRO, S. D. SCHMIDT, K. WANG, A. C. CROWLEY, Y. H. FU, S. Y. GUENETTE, D. GALAS, E. NEMENS, E. M. WIJSMAN, T. D. BIRD, G. D. SCHELLENBERG et R. E. TANZI. 1995. « Candidate gene for the chromosome 1 familial Alzheimer’s disease locus », Science, 269 : 973-977.
- MCKHANN, G., D. DRACHMAN, M. FOLSTEIN, R. KATZMAN, D. PRICE et E. M. STADLAN. 1984. « Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS‑ADRDA work group under the auspices of Department of Health and Human Services task force on Alzheimer’s disease », Neurology, 34 : 939-944.
- MESSIER, C. 2003. « Diabetes, Alzheimer’s disease and apolipoprotein genotype », Experimental Gerontology, 38 :941-946.
- PAYAMI, H., K. MONTEE, H. GRIMSLID, S. SHATTUC et J. KAYE 1996a. « Increased risk of familial late-onset Alzheimer’s disease in women », Neurology, 46 : 126-129.
- PAYAMI, H., S. ZAREPARSI, K. R. MONTEE, G. J. SEXTON, J. A. KAYE, T. D. BIRD, C. E. YU, E. M. WIJSMANN, L. L. HESTON, M. LITT et G. SHELLENBERG. 1996b. « Gender difference in apolipoprotein E-associated risk for familial Alzheimer disease: A possible clue to the higher incidence of Alzheimer disease in women », American Journal of Human Genetics, 58 : 803-811.
- PTOK, U., K. BARKOW et R. HEUN. 2002. « Fertility and number of children in patients with Alzheimer’s disease », Archives of Women’s Mental Health, 5 : 83-86.
- PTOK, U., A. PAPASSOTIROPOULOS, W. MAIER et R. HEUN. 2000. « Advanced parental age: A risk factor for Alzheimer’s disease or depression in the elderly? », International Psychogeriatrics, 12, 4 : 445-451.
- PRINCE, M., M. CULLEN et A. MANN. 1994. « Risk factors for Alzheimer’s disease and dementia: A case-control study based on the MRC elderly hypertension trial », Neurology, 44 : 97-104.
- PUGLIELLI, L., R. E.TANZI et D. M. KOVACS. 2003. « Alzheimer’s disease: The cholesterol connection ». Nature Neuroscience, 6 : 345-351.
- RAO, V. S., A. CUPPLES, C. M. VAN DUIJN, A. KURZ, R. C. GREEN, H. CHUI, R. DUARA, S. A. AUERBACH, L. VOLICER, J. WELLS, C. VAN BROECKHOVEN, J. H. GROWDON, J. L. HAINES et L. A. FARRER. 1996. « Evidence for major gene inheritance of Alzheimer disease in families of patients with and without apolipoprotein E ε4 », American Journal of Human Genetics, 59 : 664-75.
- RITCHIE, K., et S. LOVESTONE. 2002. « The dementias », The Lancet, 360 : 1759-1766.
- ROCCA, W. A., C. M. VAN DUIJN, D. CLAYTON, V. CHANDRA, L. FRATIGLIONI, A. B. GRAVES, A. HEYMAN, A. F. JORM, E. KOKMEN, K. KONDO, J. A. MORTIMER, S. L. SHALAT, H. SOININEN et A. HOFMAN. 1991. « Maternal age and Alzheimer’s disease: A collaborative re-analysis of case-control studies », International Journal of Epidemiology, 20 : S21-S27.
- ROCCHI, A., S. PELLIGRINI, G. SICILIANO et L. MURRI 2003. « Causative and susceptibility genes for Alzheimer’s disease: A review », Brain Research Bulletin, 61 : 1-24.
- RUITENBERG, A., A. OTT, J. C. VAN SWIETEN, A, HOFMAN, M. M. BRETELER. 2001. « Incidence of dementia: Does gender make a difference? », Neurobiology of Aging, 22 : 575-580.
- SAUNDERS, A. M., W. J. STRITTMATTER, D. SCHMECHEL, P. H. ST. GEORGE-HYSLOP, M. A. PERICAK-VANCE, S. H. JOO, B. L. ROSI, J. F. GUSELLA, D. R. CRAPPER‑MACLACHLAN, M. J. ALBERTS, C. HULETTE, B. CRAIN, D. GOLDBAGER et A. D. ROSES. 1993. « Association of apolipoprotein E allele ε4 with late-onset familial and sporadic Alzheimer’s disease », Neurology, 43 : 1467-1472.
- SCHUPF, N., D. KAPELL, J. H. LEE, R. OTTMAN et R. MAYEUX. 1994. « Increased risk of Alzheimer’s disease in mothers of adults with Down’s syndrome », The Lancet, 344 : 353-356.
- SHERRINGTON, R., E. I. ROGAEV, Y. LIANG, E. A. ROGAEVA, G. LEVESQUE, M. KEDA, H. CHI, C. LIN, G. LI, K. HOLMAN, T. TSUDA, L. MAR, J.-F. FONCIN, A. C. BRUNI, M. P. MONTESI, S. SORBI, I. RAINERO, L. PINESSI, L. NEE, Y. CHUMAKOV, D. POLLEN, W. WASCO, J. L. HAINES, R. DA SILVA, M. PERICAK-VANCE, R. E. TANZI, A. D. ROSES, P. E. FRASER, J. M. ROMMENS et P. H. ST. GEORGE-HYSLOP. 1995. « Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease », Nature, 375 : 754-760.
- STRITTMATTER, W. J., A. M. SAUNDERS, D. SCHMECHEL, M. A. PERICAK-VANCE, J. ENGHILD, G. S. SALVESEN et A. D. ROSES. 1993. « Apolipoprotein E: high -avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer’s disease », Proceedings of the National Academy of Sciences, USA, 90 : 1977-1981.
- THE CANADIAN STUDY OF HEALTH AND AGING. 1994. « The Canadian Study of Health and Aging: Risk factors for Alzheimer’s disease in Canada », Neurology, 44 : 2073-2080.
- TIBERGHIEN, D., Y. ROBITAILLE, A. LAROCHE-CHOLETTE, L. HOUDE, M. GRENON et D. GAUVREAU. 1993. « Retrospective assesment of relative risks of coronary arteriosclerosis and myocardial infarct in autopsy confirmed dementias of the AD and non-AD types », dans B. CORAIN, K. IQBAL, M. NICOLINI, B. WINBLAD, H. WISNIEWSKI et P. ZATTA, éd. Alzheimer’s Disease: Advances in Clinical And Basic Research. New York, John Wiley & Sons : 121-127.
- VAN DUIJN, C. M. 1996. « Epidemiology of the dementias: Recent developments and new approaches », Journal of Neurology, Neurosurgery and Psychiatry, 60 : 478-488.
- VAN DUIJN, C. M., D. CLAYTON, V. CHANDRA, L. FRATIGLIONI, A. B. GRAVES, A. HEYMAN, A. F. JORM, E. KOKMEN, K. KONDO, J. A. MORTIMER, W. A. ROCCA, S. L. SHALAT, H. SOININEN et A. HOFMAN. 1991. « Familial aggregation of Alzheimer’s disease and related disorders: A collaborative re-analysis of case-control studies », International Journal of Epidemiology, 20 : S13-S20.
- WHALLEY, L. J., A. D. CAROTHERS, S. COLLYER, R. DE MEY et A. FRACKIEWICZ. 1982. « A study of familial factors in Alzheimer’s disease », British Journal of Psychiatry, 140 : 249-256.
- WHALLEY, L. J., B. M. THOMAS et G. et J. M. STARR. 1995. « Epidemiology of presenile Alzheimer’s disease in Scotland (1974-88). II. Exposures to possible risk factors », British Journal of Psychiatry, 167 : 732-738.
- WHITE, J. A., M. MCGUE et L. L. HESTON. 1986. « Fertility and parental age in Alzheimer disease », Journal of Gerontology, 41 : 40-43.
List of tables
Tableau 1
Description de l’échantillon
Moyenne ± écart type (étendue)
Les allèles correspondent aux différentes variantes d’un gène.
*** : pas d’âge aux premiers symptômes et au décès.
Tableau 2
Nombre et dates des événements démographiques retracés dans le fichier BALSAC chez les cas et chez les témoins
Tableau 3
Âge parental et rang de naissance chez les cas et les témoins
Tableau 4
Fécondité dans les familles d’origine des cas et des témoins
0,005 < p ≤ 0,05.
Table 5
Fécondité des cas et des témoins
0,005 < p ≤ 0,05.
Tableau 6
Taux (‰) de mortalité infantile dans les fratries des cas et des témoins
Note : * 0,005 < p ≤ 0,05, ** p ≤ 0,005.
Tableau 7
Taux (‰) de mortalité infantile chez les enfants des cas et des témoins
0,005 < p ≤ 0,05.
Table 8
Âge au décès des parents des cas et des témoins