Abstracts
Résumé
Le syndrome lymphoprolifératif avec auto-immunité (ALPS, auto-immune lymphoproliferative syndrome) a été décrit il y a plus de 30 ans. Ce syndrome tumoral bénin, d’apparition précoce (en moyenne avant cinq ans), associe des adénopathies multifocales, une splénomégalie et, éventuellement, une hépatomégalie. Cette lymphoprolifération chronique s’accompagne d’une hypergammaglobulinémie, essentiellement G et A, et se caractérise par l’accumulation dans le sang et les organes lymphoïdes secondaires d’une population polyclonale de lymphocytes Tαβ matures n’exprimant ni CD4 ni CD8, appelés lymphocytes T « doubles négatifs » (LTαβ DN). Des manifestations auto-immunes sont retrouvées chez plus de deux tiers des patients ALPS, le plus souvent sous la forme de cytopénies auto-immunes (anémie hémolytique, thrombopénie et neutropénie). Des atteintes d’organes ou de systèmes (glomérulonéphrites, hépatites, uvéites, syndrome de Guillain-Barré) sont plus rarement observées. Cet article fait le point sur les défauts de la voie de signalisation Fas mis en évidence dans la genèse de l’ALPS.
Summary
Control of lymphocyte homeostasis is essential to ensure efficient immune responses and to prevent autoimmunity. Expansion followed by contraction of the lymphocyte pool are the basis of adaptive immune responses, and apoptosis is a crucial cellular modus operandi of the contraction phase. The death receptor Fas is a key player in lymphocyte apoptosis induction and patients lacking a functional Fas receptor develop a chronic lymphoproliferation termed autoimmune lymphoproliferative syndrome (ALPS). In rare instances, defects of the Fas signaling pathway have been associated with the ALPS condition. Although these defects with familial history are usually caused by inherited mutations of the corresponding genes, somatic mosaicism of these Fas mutations were also found in sporadic cases of ALPS. These findings might have important implications in deciphering the pathophysiological bases of other autoimmune diseases.
Article body
Fondements fonctionnels et cellulaires de l’ALPS
Environ la moitié des patients atteints d’ALPS, c’est-à-dire présentant au moins deux des critères suivants : syndrome tumoral, auto-immunité, proportion élevée de LTαβ DN, hypergammaglobulinémie G ou A (hyper IgG ou IgA), sont porteurs de mutations hétérozygotes du gène Fas (ALPS-I) [1-3]. Fas est un récepteur inducteur d’apoptose appartenant à la superfamille du tumor necrosis factor receptor (TNF-R) [4]. Également appelé CD95, Apo-1 ou TNFRSF6 dans la nouvelle nomenclature de la famille des TNF-R, il est exprimé sous forme d’homotrimères à la surface de nombreux types cellulaires, notamment des lymphocytes T activés. Après interaction de Fas avec son ligand naturel, FasL, le recrutement d’un complexe multimoléculaire sous-membranaire, dénommé DISC (death inducing signaling complex) [5], est déclenché par le jeu d’interactions homotypiques de domaines fonctionnels comme les domaines de mort (DD, death domain) et les domaines effecteurs de mort (DED, death effector domain) (Figure 1). Un DD intact de Fas est essentiel pour la formation du DISC, dans lequel les procaspases 8 et 10 sont recrutées, puis activées. La cascade d’activation des caspases qui en découle conduit alors la cellule vers l’apoptose. Les mutations hétérozygotes de Fas sont associées à des défauts d’apoptose, car elles conduisent à l’expression de molécules mutantes qui bloquent la formation du DISC soit par défaut d’interaction avec l’adaptateur FADD (Fas associated death domain), soit par défaut d’interaction avec FasL et absence d’oligomérisation.
Figure 1
Signalisation de l’apoptose lymphocytaire par Fas.

Après interaction Fas-FasL, le domaine de mort (DD) de Fas peut interagir avec celui de FADD (Fas associated death domain). Le domaine effecteur de mort (DED) de FADD permet le recrutement des procaspases 8 (Flice-1) et 10 (Flice-2) qui contiennent chacune deux DED dans leur prodomaine. La formation du DISC (death inducing signaling complex) entraîne le clivage des procaspases en caspases actives (hétérotétramères des petits et grands domaines p10 et p20). Cette activation peut être bloquée par des concentrations élevées d’un analogue inactif de caspase 8, Flip (Flice inhibitory protein), qui existe sous deux isoformes, l’une courte, FlipS, l’autre longue, FlipL (non représentées ici). En revanche, en présence d’une faible concentration de FlipL, les caspases 8 et 10 activent d’autres procaspases et déclenchent une cascade biochimique aboutissant à la destruction organisée de la cellule par apoptose.
La voie Fas-FasL est importante pour la régulation de l’homéostasie lymphocytaire, notamment pour éliminer les lymphocytes effecteurs après stimulation antigénique (Figure 2A). L’observation de réponses vaccinales et anti-infectieuses normales, et parallèlement de manifestations auto-immunes chez les patients présentant un ALPS suppose que l’action de la voie Fas-FasL est particulièrement importante pour réguler les proliférations induites par des antigènes du soi (Figure 2B) [2]. Il est donc possible que les patients atteints d’ALPS ne présentant pas d’auto-immunité au moment du diagnostic puissent en développer une plus tard. La raison pour laquelle cette auto-immunité est essentiellement dirigée contre les cellules d’origine hématopoïétique reste une énigme. La production des LTαβ DN et l’apparition de l’hypergammaglobulinémie (Figure 2) ont surtout été observées dans les modèles murins de déficiences en Fas ou FasL [6]. Les LTαβ DN proviendraient en grande majorité de lymphocytes T CD8 autoréactifs, ou « mal sélectionnés », qui éteignent l’expression du CD8 et ne peuvent plus être éliminés par la voie Fas [7].
Figure 2
Élimination des lymphocytes autoréactifs par Fas-FasL après activation.

Figure 2 (continuation)
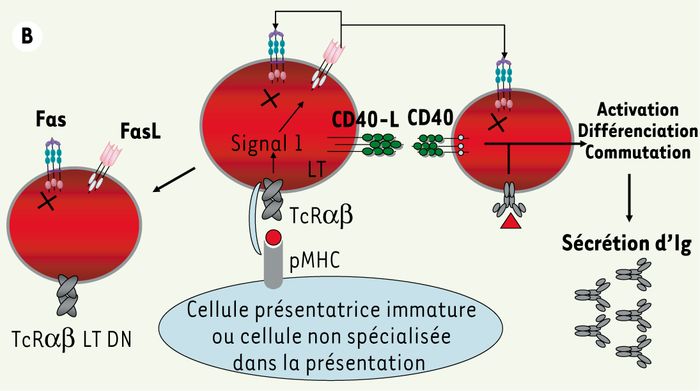
A. Les lymphocytes T recevant deux signaux d’activation (via le récepteur de l’antigène et via des molécules de costimulation comme CD28) sont insensibles à l’apoptose induite par Fas, notamment parce que l’inhibiteur Flip est fortement exprimé. Lorsque la stimulation est fournie par des cellules normalement non spécialisées dans l’activation lymphocytaire (cellules de tissus non hématopoïétiques), ou par des cellules présentatrices immatures, la voie Fas est active. Lorsque cette stimulation est chronique, FasL est exprimé, et entraîne l’élimination des lymphocytes T ou B exprimant Fas. B. Dans un contexte de déficience en Fas, le lymphocyte T qui « attend » un signal d’apoptose module son corécepteur CD4 ou CD8 (après extinction génique) et devient double-négatif. De surcroît, les lymphocytes T activés stimulent la différenciation des lymphocytes B en plasmocytes, entraînant la sécrétion d’une grande quantité d’anticorps dans le sérum (hypergammaglobulinémie).
Ces LTαβ DN sont polyclonaux, mais l’étendue précise de leur diversité n’a pas été déterminée. Cependant, l‘absence de récepteurs semi-invariants les distingue des lymphocytes NKT qui peuvent aussi être DN. Par ailleurs, l’activation des lymphocytes T in vivo conduit à l’expression de molécules d’activation impliquées dans la collaboration entre lymphocytes T et B, comme le ligand de la molécule CD40. Les lymphocytes B, stimulés par la voie CD40 et le récepteur de l’antigène, subissent alors la commutation de classe isotypique ainsi qu’une maturation en plasmocytes. En conséquence, de grandes quantités d’immunoglobulines d’isotype G et A sont sécrétées (Figure 2B).
Dans quelques rares familles consanguines, l’ALPS est associé à un défaut d’expression de Fas (voir ci-dessous). Ces patients, équivalents à des invalidations naturelles de Fas et appelés ALPS-0, présentent une lymphoprolifération très sévère qui, dans certains cas, ne peut être corrigée qu’après transplantation médullaire [8, 9]. Au-delà de l’intérêt thérapeutique, cette observation suggère, dans les limites d’un recul de 8 ans, que les conséquences d’un défaut complet de Fas chez l’homme sont restreintes au système hématopoïétique, et se manifestent par une lymphoprolifération chronique des lymphocytes T. La prolifération et la différenciation des lymphocytes B, ainsi que les manifestations auto-immunes, semblent secondaires à la lymphoprolifération T et pourraient être associées, en plus du défaut de Fas, à des défauts d’autres gènes modificateurs.
Fondements génétiques de l’ALPS associé à un défaut de Fas
Plus de 100 mutations hétérozygotes dominantes de Fas sont aujourd’hui décrites dans la littérature [10-14]. Alors que les lymphocytes de tous les porteurs de mutations héritées de Fas présentent un défaut d’apoptose in vitro, seulement 70 % des individus porteurs de ces mutations développent un ALPS, indiquant qu’au-delà du défaut de Fas d’autres facteurs sont nécessaires au déclenchement de la pathologie [10]. Dans le modèle animal correspondant, la lignée de souris lpr, l’expression phénotypique varie en fonction du fonds génétique, suggérant l’implication de gènes modificateurs [15]. L’amplitude du défaut fonctionnel d’apoptose observé chez les patients et leurs parents porteurs sains étant similaire, l’hypothèse actuelle suppose que ces gènes modificateurs ne codent pas pour des molécules impliquées dans la signalisation de l’apoptose par Fas. La limite de cette hypothèse repose sur la pertinence physiologique du test fonctionnel réalisé in vitro.
Défauts de la voie de signalisation
Des mutations de la caspase 10 ont été décrites chez trois patients (ALPS-II) au sein de deux familles [16]. Le caractère dominant ou récessif de ces défauts reste débattu [17]. Aucune validation dans un modèle animal n’a été effectuée, car l’orthologue murin de la caspase 10 n’est pas fonctionnel. Les cellules déficientes en caspase 10 sont résistantes à l’apoptose induite par Fas, ainsi que par les autres récepteurs inducteurs d’apoptose comme les TRAIL (TNF-related-apoptosis-inducing-ligand)-R.
Un défaut récessif de la caspase 8 a également été mis en évidence chez l’homme [18]. Comme attendu, les lymphocytes de ces patients sont résistants à l’apoptose induite par Fas. Cependant, les deux patients décrits, issus d’une même famille consanguine, ont développé un déficit immunitaire plutôt qu’un ALPS. Cette observation est en accord avec la description récente de l’implication de la caspase 8 et du FADD dans la prolifération lymphocytaire [19]. De même, les modèles d’invalidation lymphoïde de ces gènes ont montré une perturbation du développement lymphocytaire [20]. Chez l’homme, il semble donc que la caspase 8 ait une fonction associée à la signalisation de l’apoptose par les récepteurs de la famille du TNF-R, ainsi qu’une fonction de signalisation de la prolifération par les récepteurs de l’antigène comme le TCR, le BCR et le récepteur Fc des immunoglobulines. La caspase 10, quant à elle, semble spécialisée dans la signalisation de l’apoptose par des récepteurs tels que Fas ou les TRAIL-R. L’ALPS peut donc être la conséquence d’un défaut de Fas complet (ALPS-0) ou partiel (ALPS-I), ou plus rarement d’un défaut de caspase 10 (ALPS-II) (Figure 3).
Figure 3
Défauts de Fas et de sa voie de signalisation.

Les défauts complets d’expression de Fas (ALPS-0) sont associés à des mutations homozygotes de Fas et à un syndrome lymphoprolifératif très sévère. Les mutations hétérozygotes de Fas (ALPS-I) entraînent le plus souvent une expression normale de Fas. La protéine mutante (ici tronquée, sans DD) exerce un effet dominant négatif sur la protéine sauvage en inhibant la formation du DISC. Les défauts de caspase 10 (ALPS-II) entraînent également une inhibition de l’activation de la cascade biochimique aboutissant normalement à l’apoptose. Les mutations de la caspase 8 sont associées à des défauts de prolifération lymphocytaire, un défaut associé à un déficit immunitaire combiné (des lymphocytes T, B et NK) modéré.
Fondements génétiques des formes sporadiques d’ALPS
L’apoptose induite par Fas mesurée in vitro est normale dans les lymphocytes de la moitié des patients atteints d’ALPS, excluant a priori un défaut de Fas. La plupart de ces patients correspondent à des cas sporadiques d’ALPS, qui ne présentent souvent que 2 des 4 critères d’inclusion. L’étude des patients pour lesquels un pourcentage important de lymphocytes T DN est retrouvé dans le sang a permis d’identifier des mutations de Fas dans cette population lymphocytaire. L’analyse détaillée des différentes lignées hématopoïétiques a montré la présence de ces mutations dans 10 % à 20 % des granulocytes, des monocytes et des différentes populations lymphocytaires. Les LTαβ DN, phénotypiquement et fonctionnellement identiques à ceux observés dans les formes héritées d’ALPS, sont tous mutés. Ces mutations sont indétectables sur des cellules du bulbe capillaire ou de la muqueuse buccale, indiquant qu’il s’agit de mutations somatiques qui sont probablement apparues au cours du développement au sein d’un précurseur hématopoïétique [21]. Chez un patient, l’analyse d’une population enrichie en progéniteurs hématopoïétiques purifiés à partir de la rate a montré qu’environ 1 % de ces cellules étaient mutées (Figure 4). Des mutations somatiques de Fas ont maintenant été identifiées chez dix patients ALPS, qui sont donc des mosaïques. Paradoxalement, alors que les lymphocytes T mutés sont résistants à l’apoptose induite par Fas, ils disparaissent en culture in vitro, qu’ils soient stimulés ou non par des mitogènes ou des superantigènes. En conséquence, le test fonctionnel, qui ne peut être réalisé que sur des cellules activées en culture, est normal.
Figure 4
Ontogénie des cellules mutantes chez les patients mosaïques (ALPS-Im).

Les mutations identifiées dans les cellules sanguines de patients mosaïques sont retrouvées dans 1 % à 2 % des cellules souches hématopoïétiques. En périphérie, dans le sang les ganglions lymphatiques et la rate, les mutations sont retrouvées dans 10 % à 20 % (selon les patients) des cellules myéloïdes et lymphoïdes, suggérant que 10 % à 20 % des progéniteurs communs sont également mutés. Fas pourrait être un régulateur de l’homéostasie de progéniteurs hématopoïétiques. L’accumulation de cellules mutées dans la population de lymphocytes Tαβ DN indique que les stimulations qui conduisent à leur apparition sont strictement régulées par Fas. La stimulation in vitro des lymphocytes de patients mosaïques conduit à l’accumulation de cellules non mutées. La disparition des cellules mutées peut s’expliquer par l’activation compensatrice d’autres voies d’apoptose, ou par un défaut de facteurs de survie ou de prolifération, absent dans les conditions de culture.
La description de patients mosaïques pour des mutations hétérozygotes de Fas conduit aux conclusions suivantes. D’une part, une proportion limitée de cellules mutées est suffisante pour déclencher un syndrome lymphoprolifératif cliniquement identique à celui de patients dont la mutation héritée est présente dans toutes les cellules. D’autre part, les mutations somatiques confèrent un avantage sélectif aux cellules mutées in vivo, indiquant que ces événements génétiques peuvent également intervenir dans des cellules non tumorales et non transformées. L’enrichissement de cellules mutantes observé entre la population de progéniteurs hématopoïétiques et les leucocytes sanguins suggère que Fas est impliquée dans le contrôle du développement lymphoïde et myéloïde. De même, la présence des mutations dans 100 % des LTαβ DN indique que les stimulations qui conduisent à l’apparition de cette population polyclonale sont strictement régulées par Fas. L’identification des cibles reconnues par ces lymphocytes devrait apporter des informations sur la nature des stimuli conduisant à l’ALPS. Leur apoptose spontanée en culture est à ce jour une limite incontournable qui grève toute étude fonctionnelle. Enfin, les mutations somatiques représentent environ 15 % des mutations de Fas dans l’ALPS. Compte tenu de la fréquence de ces événements, il est possible que des mutations somatiques d’autres gènes impliqués dans l’apoptose constituent le second facteur associé au déclenchement de l’ALPS. De telles mutations somatiques pourraient également être impliquées dans d’autres maladies auto-immunes sporadiques, ou pourraient expliquer les variations d’expression phénotypique de telles maladies observées parfois chez les jumeaux monozygotes. La présence d’une population lymphocytaire pathognomonique (spécifique de la pathologie étudiée) comme les LTαβ DN a permis de mettre en évidence ces mutations somatiques dans les formes sporadiques d’ALPS. La mise en évidence de populations lymphocytaires spécifiques d’autres maladies auto-immunes sporadiques pourraient ainsi ouvrir de nouvelles perspectives pour identifier le ou les facteurs génétiques impliqués.
Conclusions
L’identification de mutations héritées du gène Fas a permis d’ébaucher le schéma de la physiopathologie de l’ALPS. La description de défauts de la voie de signalisation de Fas a confirmé le rôle central de cette voie d’apoptose dans la pathologie. Enfin, la mise en évidence récente de mutations somatiques de Fas dans des cas sporadiques d’ALPS a révélé l’importance de la population de LTαβ DN. Cette dernière découverte pourrait avoir des implications importantes pour la compréhension d’autres maladies auto-immunes.
Appendices
Références
- 1. Canale VC, Smith CH. Chronic lymphadenopathy simulating malignant lymphoma. J Pediatr 1967 ; 70 : 891-9.
- 2. Rieux-Laucat F, Fischer A, Deist FL. Cell-death signaling and human disease. Curr Opin Immunol 2003 ; 15 : 325-31.
- 3. Rieux-Laucat F, Le Deist F, Fischer A. Autoimmune lymphoproliferative syndromes : genetic defects of apoptosis pathways. Cell Death Differ 2003 ; 10 : 124-33.
- 4. Lavrik I, Golks A, Krammer PH. Death receptor signaling. J Cell Sci 2005 ; 118 : 265-7.
- 5. Peter ME, Krammer PH. The CD95 (APO-1/Fas) DISC and beyond. Cell Death Differ 2003 ; 10 :26-35.
- 6. Nagata S, Suda T. Fas and Fas ligand : lpr and gld mutations. Immunol Today 1995 ; 16 : 39-43.
- 7. Pestano GA, Zhou Y, Trimble LA, et al. Inactivation of misselected CD8 T cells by CD8 gene methylation and cell death. Science 1999 ; 284 : 1187-91.
- 8. Rieux-Laucat F, Le Deist F, Hivroz C, et al. Mutations in fas associated with human lymphoproliferative syndrome and autoimmunity. Science 1995 ; 268 : 1347-9.
- 9. Benkerrou M, Le Deist F, de Villartay J, et al. Correction of fas (CD95) deficiency by haploidentical bone marrow transplantation. Eur J Immunol 1997 ; 27 : 2043-7.
- 10. Rieux-Laucat F, Blachere S, Danielan S, et al. Lymphoproliferative syndrome with autoimmunity : a possible genetic basis for dominant expression of the clinical manifestations. Blood 1999 ; 94 : 2575-82.
- 11. Vaishnaw AK, Orlinick JR, Chu JL, et al. The molecular basis for apoptotic defects in patients with CD95 (Fas/Apo-1) mutations. J Clin Invest 1999 ; 103 : 355-63.
- 12. Bleesing JJ, Brown MR, Straus SE, et al. Immunophenotypic profiles in families with autoimmune lymphoproliferative syndrome. Blood 2001 ; 98 : 2466-73.
- 13. Drappa J, Vaishnaw AK, Sullivan KE, et al. Fas gene mutations in the Canale-Smith syndrome, an inherited lymphoproliferative disorder associated with autoimmunity. N Engl J Med 1996 ; 335 : 1643-9.
- 14. Jackson CE, Fischer RE, Hsu AP, et al. Autoimmune lymphoproliferative syndrome with defective Fas : genotype influences penetrance. Am J Hum Genet 1999 ; 64 : 1002-14.
- 15. Morel L, Tian XH, Croker BP, Wakeland EK. Epistatic modifiers of autoimmunity in a murine model of lupus nephritis. Immunity 1999 ; 11 : 131-9.
- 16. Wang J, Zheng L, Lobito A, et al. Inherited human Caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type II. Cell 1999 ; 98 : 47-58.
- 17. Gronbaek K, Dalby T, Zeuthen J, et al. The V410I (G1228A) variant of the caspase-10 gene is a common polymorphism of the Danish population. Blood 2000 ; 95 : 2184-5.
- 18. Chun HJ, Zheng L, Ahmad M, et al. Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature 2002 ; 419 : 395-9.
- 19. Su H, Bidere N, Zheng L, et al. Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science 2005 ; 307 : 1465-8.
- 20. Salmena L, Lemmers B, Hakem A, et al. Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev 2003 ; 17 : 883-95.
- 21. Holzelova E, Vonarbourg C, Stolzenberg MC, et al. Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N Engl J Med 2004 ; 351 : 1409-18.
List of figures
Figure 1
Signalisation de l’apoptose lymphocytaire par Fas.
Après interaction Fas-FasL, le domaine de mort (DD) de Fas peut interagir avec celui de FADD (Fas associated death domain). Le domaine effecteur de mort (DED) de FADD permet le recrutement des procaspases 8 (Flice-1) et 10 (Flice-2) qui contiennent chacune deux DED dans leur prodomaine. La formation du DISC (death inducing signaling complex) entraîne le clivage des procaspases en caspases actives (hétérotétramères des petits et grands domaines p10 et p20). Cette activation peut être bloquée par des concentrations élevées d’un analogue inactif de caspase 8, Flip (Flice inhibitory protein), qui existe sous deux isoformes, l’une courte, FlipS, l’autre longue, FlipL (non représentées ici). En revanche, en présence d’une faible concentration de FlipL, les caspases 8 et 10 activent d’autres procaspases et déclenchent une cascade biochimique aboutissant à la destruction organisée de la cellule par apoptose.
Figure 2
Élimination des lymphocytes autoréactifs par Fas-FasL après activation.
Figure 2 (continuation)
A. Les lymphocytes T recevant deux signaux d’activation (via le récepteur de l’antigène et via des molécules de costimulation comme CD28) sont insensibles à l’apoptose induite par Fas, notamment parce que l’inhibiteur Flip est fortement exprimé. Lorsque la stimulation est fournie par des cellules normalement non spécialisées dans l’activation lymphocytaire (cellules de tissus non hématopoïétiques), ou par des cellules présentatrices immatures, la voie Fas est active. Lorsque cette stimulation est chronique, FasL est exprimé, et entraîne l’élimination des lymphocytes T ou B exprimant Fas. B. Dans un contexte de déficience en Fas, le lymphocyte T qui « attend » un signal d’apoptose module son corécepteur CD4 ou CD8 (après extinction génique) et devient double-négatif. De surcroît, les lymphocytes T activés stimulent la différenciation des lymphocytes B en plasmocytes, entraînant la sécrétion d’une grande quantité d’anticorps dans le sérum (hypergammaglobulinémie).
Figure 3
Défauts de Fas et de sa voie de signalisation.
Les défauts complets d’expression de Fas (ALPS-0) sont associés à des mutations homozygotes de Fas et à un syndrome lymphoprolifératif très sévère. Les mutations hétérozygotes de Fas (ALPS-I) entraînent le plus souvent une expression normale de Fas. La protéine mutante (ici tronquée, sans DD) exerce un effet dominant négatif sur la protéine sauvage en inhibant la formation du DISC. Les défauts de caspase 10 (ALPS-II) entraînent également une inhibition de l’activation de la cascade biochimique aboutissant normalement à l’apoptose. Les mutations de la caspase 8 sont associées à des défauts de prolifération lymphocytaire, un défaut associé à un déficit immunitaire combiné (des lymphocytes T, B et NK) modéré.
Figure 4
Ontogénie des cellules mutantes chez les patients mosaïques (ALPS-Im).
Les mutations identifiées dans les cellules sanguines de patients mosaïques sont retrouvées dans 1 % à 2 % des cellules souches hématopoïétiques. En périphérie, dans le sang les ganglions lymphatiques et la rate, les mutations sont retrouvées dans 10 % à 20 % (selon les patients) des cellules myéloïdes et lymphoïdes, suggérant que 10 % à 20 % des progéniteurs communs sont également mutés. Fas pourrait être un régulateur de l’homéostasie de progéniteurs hématopoïétiques. L’accumulation de cellules mutées dans la population de lymphocytes Tαβ DN indique que les stimulations qui conduisent à leur apparition sont strictement régulées par Fas. La stimulation in vitro des lymphocytes de patients mosaïques conduit à l’accumulation de cellules non mutées. La disparition des cellules mutées peut s’expliquer par l’activation compensatrice d’autres voies d’apoptose, ou par un défaut de facteurs de survie ou de prolifération, absent dans les conditions de culture.