Article body
L’étude du syndrome de prédisposition mendélienne aux infections mycobactériennes (MIM 209950) a permis de mieux caractériser les mécanismes moléculaires de l’immunité anti-mycobactérienne chez l’homme. En effet, certains malades présentent une vulnérabilité héréditaire spécifique vis-à-vis des infections mycobactériennes [1-3]. Des études moléculaires ont montré qu’ils sont porteurs de mutations germinales dans cinq gènes participant aux voies d’activation cellulaire de l’interferon-γ (IFN-γ) ou de l’interleukine-12 (IL-12). Cela met en évidence le rôle indispensable de l’IFN-γ et de son principal inducteur l’IL-12 [4-10], dans le contrôle des infections mycobactériennes chez l’homme [11-13].
Nous avons récemment réalisé une étude sur trois enfants présentant des infections mycobactériennes disséminées chez lesquels nous avons trouvé une mutation faux sens T168N dans le gène IFNGR2. Ils sont issus de deux familles consanguines, non apparentées entre elles. La première patiente est originaire d’Iran et a développé une infection disséminée à l’âge d’un mois après vaccination par le bacille de Calmette et Guérin (BCG, c’est-à-dire M. bovis atténué). Actuellement âgée de deux ans et demi, elle est toujours traitée par des antibiotiques antimycobactériens. Les deux autres malades sont deux frères originaires d’Arabie Saoudite qui avaient développé une infection disséminée à M. fortiutum ; le premier est mort à l’âge de six ans, et le second, âgé de cinq ans, est toujours traité par des antibiotiques antimycobactériens.
Chez ces trois enfants, nous avons identifié un défaut complet de réponse à l’IFN-γ. Les explorations ont été effectuées sur sang total [14], lignées lymphocytaires B immortalisées par le virus Epstein-Barr et sur fibroblastes transformés par l’antigène T SV40 issus des patients. La même mutation homozygote T168N du gène IFNGR2 a été retrouvée chez ces trois patients. Puis, par des techniques de microscopie confocale et de biotinylation de surface cellulaire des récepteurs, nous avons démontré que la protéine IFN-γR2 T168N mutante était exprimée, comme la protéine sauvage, à la surface cellulaire. Ces résultats attestaient que le défaut de réponse n’était pas dû à une localisation cellulaire anormale ou à une absence d’expression en surface. De plus, nous avons découvert que la mutation T168N avait un poids moléculaire apparent beaucoup plus élevé que celui de la forme sauvage. Ce gain de poids moléculaire ne semblait pas correspondre au seul changement d’acide aminé causé par la mutation. En effet, la mutation crée un nouveau site consensus de glycosylation (TSTA → NSTA) dans la chaîne 2 du récepteur de l’IFN-γ (IFN-γR2) [15]. Jusqu’à présent les mutations entraînant un gain de glycosylation ont toujours été considérées comme rares.
Par l’utilisation de glucosidase (PNGaseF) ou d’inhibiteur chimique de la glycosylation (Tunicamycine), nous avons ensuite formellement démontré que le gain de poids était exclusivement dû à un gain de glycosylation. Puis, par mutagenèse dirigée, nous avons montré que la nouvelle molécule d’hydrate de carbone ajoutée était en elle-même délétère, abolissant la fonction du récepteur de l’IFN-γ. La complémentation du phénotype cellulaire a été effectuée in vitro et ex vivo, en traitant les cellules des patients par un inhibiteur de la N-glycosylation (Tunicamycine) ou par le PNGaseF (Figure 1). Nous avons ainsi prouvé qu’il était possible d’effectuer une complémentation chimique, sur les cellules du patient, avec des concentrations de drogues permettant une réponse normale en terme de synthèse protéique. Ces expériences démontrent que l’ajout de cet hydrate de carbone est directement responsable du défaut de réponse à l’IFN-γ.
Une proportion importante de mutations responsables de maladies génétiques humaines pourrait donc être liée à la création de nouveaux sites de glycosylation. Ces mutations créent un « gain de glycosylation » et définissent un nouveau groupe de maladies génétiques, ouvrant de nouvelles possibilités thérapeutiques grâce à l’utilisation d’inhibiteurs de la glycosylation [16]. Pour les rechercher, nous avons ensuite criblé in silico la banque Human Gene Mutation Database (http://www.hgmd.org/) afin d’identifier des mutations faux-sens pouvant entraîner un gain de glycosylation. À partir de 10 047 mutations dans 577 gènes codant pour des protéines circulant à travers la voie sécrétoire, nous avons trouvé 142 mutations candidates (~1,4 %) dans 77 gènes (~13,3 %). Une proportion importante de mutations causales dans les maladies génétiques humaines semble donc être liée à la création de nouveaux sites de glycosylation [16]. Le nombre de mutations responsables d’un gain de glycosylation est très significativement augmenté par rapport au nombre attendu du fait du hasard (p = 2.10-7) (Tableau I). Quatorze de ces gènes ainsi déterminés sont associés à des immunodéficiences primaires. En sélectionnant au hasard six mutations dans cinq gènes (CD18, IL2RG, CD8A, CD40LG et PRF1) codant des protéines qui transitent par le réticulum endoplasmique, les six protéines mutantes choisies au hasard dans les immuno-déficiences primaires se sont avérées toutes correspondre à un gain de glycosylation. En étudiant deux de ces mutation dans les gènes CD18 et CD40LG, nous avons formellement établi par des études fonctionnelles que leur pathogénicité était lié au gain de glycosylation, démontrant l’universalité de notre hypothèse.
Tableau I
Un total de 20 667 mutations faux-sens pathogènes comprises dans 1 325 gènes ont été analysées dans la HGMD (Human Gene Mutation Database) (mise à jour de mai 2004).
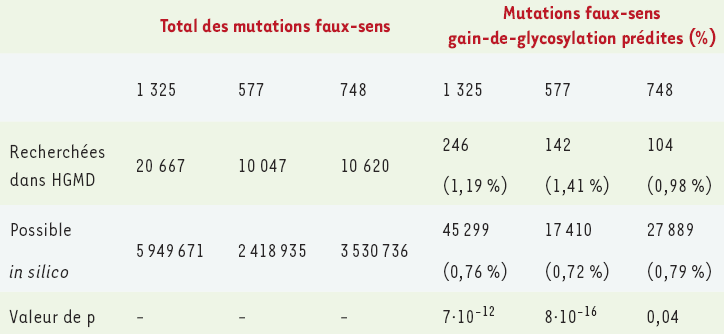
Parmi ces gènes, 577 codent pour des protéines qui sont prédites pour entrer dans le réticulum endoplasmique et ainsi être potentiellement glycosylées (fondé sur l’identification, in silico, des peptides signaux ou d’ancrages) avec une probabilité combinée supérieure à 0,75. Les 748 gènes restants sont ceux qui ne sont pas prédits pour entrer dans le réticulum endoplasmique. De ces groupes de 1 325, 577 et 748 gènes, les mutations dans 158, 77 et 81 gènes, respectivement, causent potentiellement des gains de glycosylation. Pour chacun de ces trois groupes de gènes, le nombre de mutations faux-sens pathogènes documentées, entraînant un gain de glycosylation prédit a été comparé avec le nombre total de mutations faux-sens possibles (créées in silico) pouvant créer un site consensus de glycosylation. Une statistique de χ2 a ensuite été utilisée pour comparer les proportions observées avec celles attendues (valeurs de p indiquées) (adapté de [16]).
Figure 1
Complémentation chimique du phénotype cellulaire.

Des fibroblastes transformés par l’antigène T SV40 d’un contrôle positif (C+) d’un des patients portant la mutation T168N (P2) et d’un contrôle négatif portant l’allèle 278delAG IFNGR2 (C-) [5] ont été incubés 48 heures dans un millieu de culture complet avec (histogramme rouge) ou sans (histogramme jaune) IFNγ (105 IU.ml-1), tunicamycine (0,1 µg.ml-1) ou PNGase-F (750 IU.ml-1). L’expression de surface des molécules HLA-DR a été déterminée par la technique de cytométrie en flux en utilisant un anticorps spécifique directement couplé à un fluorochrome (adapté de [16]).
En conclusion
L’ajout d’une nouvelle molécule d’hydrate de carbone est responsable du phénotype cellulaire de défaut de réponse à l’IFN-γ chez les patients porteurs de la mutation T168N. De rares mutations germinales responsables d’un gain de glycosylation avaient déjà été décrites dans la littérature [16]. Mais le caractère pathogène de ces nouveaux hydrates de carbone n’avait jamais été établi de façon expérimentale. Or, il s’avère qu’une proportion importante de mutations mendéliennes faux-sens, responsables de maladies génétiques, crée des sites de N-glycosylation qui pourraient être directement impliqués dans le processus pathogénique. Ces mutations faux-sens peuvent potentiellement être complémentées par des inhibiteurs de la glycosylation ou par des glucosidases, comme la mutation T168N du récepteur de l’interféron-γ. Nous avons ainsi identifié une nouvelle classe de mutations définissant un nouveau groupe de maladies humaines : les mutations avec gain de glycosylation. La conséquence directe de l’ajout d’une nouvelle chaîne d’hydrate de carbone, et non de la modification per se de l’acide aminé, serait en effet responsable d’un certain nombre de maladies humaines, ouvrant ainsi la voie à une thérapie chimique ciblée. Ce type de mutation doit être systématiquement recherché car les malades qui en sont porteurs pourraient bénéficier de traitements diminuant la glycosylation, offrant ainsi une alternative thérapeutique à la transplantation ou à la thérapie génique [16].
Appendices
Références
- 1. Casanova JL, Abel L. Genetic dissection of immunity to mycobacteria : the human model. Annu Rev Immunol 2002 ; 20 : 581-620.
- 2. Casanova JL, Blanche S, Emile JF, et al. Idiopathic disseminated bacillus Calmette-Guerin infection : a French national retrospective study. Pediatrics 1996 ; 98 : 774-8.
- 3. Casanova JL, Jouanguy E, Lamhamedi S, et al. Immunological conditions of children with BCG disseminated infection. Lancet 1995 ; 346 : 581.
- 4. Jouanguy E, Altare F, Lamhamedi S, et al. Interferon-gamma-receptor deficiency in an infant with fatal bacille Calmette-Guerin infection. N Engl J Med 1996 ; 335 : 1956-61.
- 5. Dorman SE, Holland SM. Mutation in the signal-transducing chain of the interferon-gamma receptor and susceptibility to mycobacterial infection. J Clin Invest 1998 ; 101 : 2364-9.
- 6. Dupuis S, Dargemont C, Fieschi C, et al. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science 2001 ; 293 : 300-3.
- 7. Fieschi C, Bosticardo M, de Beaucoudrey L, et al. A novel form of complete IL-12/IL-23 receptor beta1 deficiency with cell surface-expressed nonfunctional receptors. Blood 2004 ; 104 : 2095-101.
- 8. Jouanguy E, Lamhamedi-Cherradi S, Lammas D, et al. A human IFNGR1 small deletion hotspot associated with dominant susceptibility to mycobacterial infection. Nat Genet 1999 ; 21 : 370-8.
- 9. Picard C, Fieschi C, Altare F, et al. Inherited interleukin-12 deficiency : IL12B genotype and clinical phenotype of 13 patients from six kindreds. Am J Hum Genet 2002 ; 70 : 336-48.
- 10. Jouanguy E, Lamhamedi-Cherradi S, Altare F, et al. Partial interferon-gamma receptor 1 deficiency in a child with tuberculoid bacillus Calmette-Guerin infection and a sibling with clinical tuberculosis. J Clin Invest 1997 ; 100 : 2658-64.
- 11. Casanova JL, Abel L. The human model : a genetic dissection of immunity to infection in natural conditions. Nat Rev Immunol 2004 ; 4 : 55-66.
- 12. Casanova JL, Abel L. Inborn errors of immunity to infection : the rule rather than the exception. J Exp Med 2005 ; 202 : 197-201.
- 13. Fieschi C, Dupuis S, Catherinot E, et al. Low penetrance, broad resistance, and favorable outcome of interleukin 12 receptor beta1 deficiency : medical and immunological implications. J Exp Med 2003 ; 197 : 527-35.
- 14. Feinberg J, Fieschi C, Doffinger R, et al. Bacillus Calmette Guerin triggers the IL-12/IFN-gamma axis by an IRAK-4- and NEMO-dependent, non-cognate interaction between monocytes, NK, and T lymphocytes. Eur J Immunol 2004 ; 34 : 3276-84.
- 15. Lowe JB, Marth JD. A genetic approach to mammalian glycan function. Annu Rev Biochem 2003 ; 72 : 643-91.
- 16. Vogt G, Chapgier A, Yang K, et al. Gains of glycosylation comprise an unexpectedly large group of pathogenic mutations. Nat Genet 2005 ; 37 : 692-700.
List of figures
Figure 1
Complémentation chimique du phénotype cellulaire.
Des fibroblastes transformés par l’antigène T SV40 d’un contrôle positif (C+) d’un des patients portant la mutation T168N (P2) et d’un contrôle négatif portant l’allèle 278delAG IFNGR2 (C-) [5] ont été incubés 48 heures dans un millieu de culture complet avec (histogramme rouge) ou sans (histogramme jaune) IFNγ (105 IU.ml-1), tunicamycine (0,1 µg.ml-1) ou PNGase-F (750 IU.ml-1). L’expression de surface des molécules HLA-DR a été déterminée par la technique de cytométrie en flux en utilisant un anticorps spécifique directement couplé à un fluorochrome (adapté de [16]).
List of tables
Tableau I
Un total de 20 667 mutations faux-sens pathogènes comprises dans 1 325 gènes ont été analysées dans la HGMD (Human Gene Mutation Database) (mise à jour de mai 2004).
Parmi ces gènes, 577 codent pour des protéines qui sont prédites pour entrer dans le réticulum endoplasmique et ainsi être potentiellement glycosylées (fondé sur l’identification, in silico, des peptides signaux ou d’ancrages) avec une probabilité combinée supérieure à 0,75. Les 748 gènes restants sont ceux qui ne sont pas prédits pour entrer dans le réticulum endoplasmique. De ces groupes de 1 325, 577 et 748 gènes, les mutations dans 158, 77 et 81 gènes, respectivement, causent potentiellement des gains de glycosylation. Pour chacun de ces trois groupes de gènes, le nombre de mutations faux-sens pathogènes documentées, entraînant un gain de glycosylation prédit a été comparé avec le nombre total de mutations faux-sens possibles (créées in silico) pouvant créer un site consensus de glycosylation. Une statistique de χ2 a ensuite été utilisée pour comparer les proportions observées avec celles attendues (valeurs de p indiquées) (adapté de [16]).