Abstracts
Résumé
La matrice extracellulaire (MEC) est un assemblage de macromolécules (collagènes, protéoglycanes, élastine et glycoprotéines de structure) qui lient des cellules homologues ou hétérologues et les organisent en tissus. La MEC entoure les cellules qui la synthétisent et détermine en retour leur phénotype. Les macromolécules sont intensivement synthétisées pendant le développement et la croissance de l’individu. Chez l’adulte, leur renouvellement est lent. Au cours du vieillissement, l’interaction des macromolécules avec les facteurs environnementaux (glucose, lipides, calcium…) et l’altération des processus de synthèse et de dégradation perturbent l’homéostasie de la matrice et, en conséquence, la fonctionnalité des tissus. Tout processus pathologique accélère les altérations de la MEC, en particulier artérielle.
Summary
The extracellular matrix provides a structural framework essential for the functional properties of tissues. In each tissue, the three-dimensional organisation of the extracellular matrix molecules - elastin, collagens, proteoglycans and structural glycoproteins - synthesized during development and growth is optimal for these functions. In adult tissues, proteases are constitutively expressed but have a very low activity and the turn-over of elastic and collagen fibers is very low. During ageing, the interaction of environmental factors (glucose, lipids, calcium…) and modifications of the biosynthesis and degradation processes lead to modifications of extracellular matrix homeostasis and consequently to alterations of tissue fonctionality. These alterations are increased during pathological processes such as cardiovascular diseases.
Article body
La matrice extracellulaire (MEC) est un assemblage de macromolécules (de nature protéique et glucidique) qui lient entre elles des cellules homologues ou hétérologues et les organisent en tissus (seuls les êtres unicellulaires n’ont pas de matrice extracellulaire). La matrice extracellulaire est plus ou moins importante et de composition très différente selon le tissu considéré : le derme et les tendons sont presque exclusivement constitués de matrice extracellulaire, le cerveau en contient très peu. Les macromolécules de la matrice extracellulaire sont regroupées en quatre catégories : les collagènes, les protéoglycanes, l’élastine et les glycoprotéines de structure. La proportion de chacun de ces éléments est très variable selon le tissu considéré.
Après la description de chacune de ces familles, nous introduirons les mécanismes de vieillissement de la matrice extracellulaire. Ne pouvant, dans ce chapitre, présenter de manière exhaustive la matrice extracellulaire de tous les tissus [1], nous nous focaliserons sur la matrice extracellulaire artérielle et son vieillissement dans les conditions normales et pathologiques.
Les macromolécules de la MEC
Même si la MEC constitue une charpente dans laquelle sont intégrées les cellules, les interactions cellules-matrices sont très étroites. Les cellules synthétisent la MEC qui les entoure et cette matrice détermine en retour le phénotype des cellules [2, 3].
Collagènes
Les collagènes sont les protéines les plus représentées dans l’organisme humain [4]. Le derme, les tendons, les parois vasculaires en contiennent respectivement 65-75 %, 70-85 % et 20-40 % (% du poids sec). Les collagènes forment une famille de 27 membres distincts (I à XXVII) regroupés en deux grandes sous-familles : les collagènes fibrillaires et les collagènes non-fibrillaires. Chaque molécule de collagène est un homotrimère ou un hétérotrimère composé de trois chaînes α, ayant un ou plusieurs domaines en triple hélice. La répétition du triplet (Gly-X-Y, X étant le plus souvent une proline et Y une hydroxyproline) sur les trois chaînes α permet cette conformation en triple hélice.
Les collagènes les plus représentés sont les collagènes fibrillaires I (derme, os, tendon, ligament, artères, veines…), II (cartilage) et III (derme, artères, veines…). Lorsqu’ils sont sécrétés par les cellules, les collagènes sont des triples hélices flanquées de domaines amino et carboxyterminaux globulaires (Figure 1A). Ces domaines sont excisés avant que les triples hélices ne s’associent de façon très organisée pour former les fibres de collagènes. Les collagènes fibrillaires moins représentés comme le collagène V et les collagènes non-fibrillaires comme le collagène IV ont également des rôles essentiels. Le collagène V détermine le diamètre des fibres de collagène I auquel il est associé ; le collagène IV forme la charpente de la jonction dermo-épidermique et autres membranes basales.
Figure 1
Synthèse des collagènes fibrillaires (A) et des fibres élastiques (B).
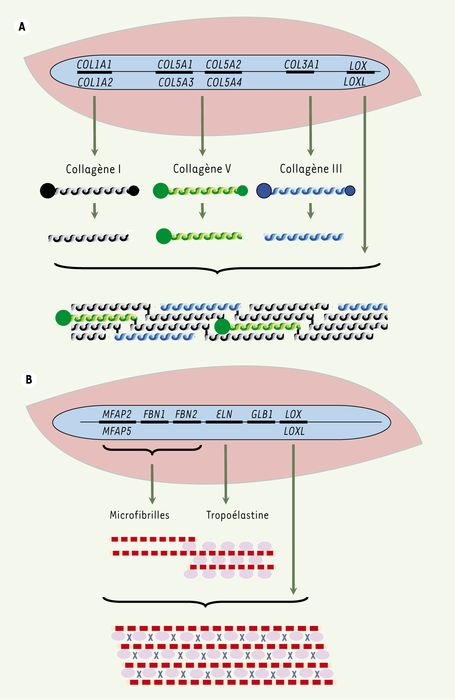
A. Chaque triple hélice de collagène résulte de l’association de trois chaînes α codées par un gène (homotrimère) ou plusieurs gènes (hétérotrimère). Les gènes COL1A1, 2, codent pour les chaînes α1 et α2 du collagène I, les gènes COL5A1, 2, 3, 4 pour celles du collagène V et le gène COL3A1 pour celles du collagène III. La triple hélice flanquée de ses extrémités globulaires amino (gros cercles pleins) et carboxyterminale (petits cercles pleins) est sécrétée dans l’espace extracellulaire. Après l’action des amino et carboxyprotéinases, les triples hélices s’associent pour former les fibrilles. Les triples hélices sont liées de manière covalente les unes aux autres lors de la formation des acides aminés de pontage, formation dont la première étape est catalysée par une lysyl-oxidase (LOX ou LOXL). B. Au cours du développement, les microfibrilles formées des glycoprotéines de structure comme les fibrillines et de MAGP (microfibrillar-associated glycoproteins) sont synthétisées et sécrétées dans l’espace extracellulaire ; elles constituent une charpente sur laquelle se déposent les molécules de tropoélastine. Une lysyl-oxydase (LOX ou LOXL) catalyse la première étape de formation des acides aminés de pontage qui résultent de la liaison de deux, trois ou quatre résidus lysine. Les acides aminés de pontage desmosine et isodesmosine sont spécifiques de l’élastine. MFAP2, 5, gène de la MAGP1, 2 ; FBN1, 2 : gène de la fibrilline 1, 2 ; ELN : gène de l’élastine ; GLB1 : gène de la β-galactosidase 1 dont un des ARNm code l’elastin-binding protein.
Une mutation sur une chaîne α de collagène est plus délétère quand elle concerne un triplet Gly-X-Y à l’extrémité carboxyterminale d’une chaîne α : des mutations des chaînes α1 ou α2 du collagène I sont responsables d’ostéogenèses imparfaites et de syndromes d’Ehlers-Danlos de type I, II, VII ; des mutations au niveau du collagène IV sont associées au syndrome d’Alport et des mutations du collagène VII à certaines formes d’épidermolyse bulleuse. La fonction principale des collagènes est de conférer une résistance mécanique aux tissus.
Protéoglycanes
Les protéoglycanes, anciennement désignés par le terme mucopolysaccharides, sont des chaînes protéiques sur lesquelles sont liées une à plusieurs dizaines de chaînes glycosaminoglycanes [5]. L’hyaluronane est un glycosaminoglycane non lié à une protéine. Longtemps désignés en fonction de la qualité des chaînes glycaniques (chondroïtine sulfate, dermatane sulfate, héparane sulfate…), les protéoglycanes sont aujourd’hui nommés selon la qualité de la chaîne protéique ou sa fonction (décorine, versicane, fibromoduline…).
De par la présence de groupes sulfatés et carboxylates, les glycosaminoglycanes sont des molécules chargées négativement et ont la capacité de fixer de nombreuses molécules d’eau. Cette propriété d’hydratation qu’ils confèrent aux tissus est essentielle à ceux d’entre eux qui subissent de fortes variations de pression comme les cartilages et les vaisseaux sanguins. Vingt à trente pour cent du poids sec des cartilages sont des glycosaminoglycanes. En fixant des facteurs de croissance comme les bFGF (basic fibroblast growth factor), des serpines comme l’antithrombine III et la protéase nexine-1, ou en jouant le rôle de co-récepteurs, les héparane sulfate et héparine contrôlent la biodisponibilité et/ou la fonction de ces facteurs.
Élastine
L’élastine est le composant majeur des fibres élastiques ; elle est associée à des microfibrilles composées de glycoprotéines de structure comme les fibrillines et les MAGP (microfibril-associated glycoproteins) [6]. Les fibres élastiques des artères sont organisées en lames concentriques parallèles à la surface du vaisseau ; elles contiennent 90 % d’élastine. Les fibres élastiques de la peau sont dénommées fibres oxytalanes, fibres d’élaunine et fibres élastiques selon les proportions d’élastine et de glycoprotéines de structure associées. L’élastine est synthétisée sous forme d’un précurseur soluble, la tropoélastine (Figure 1B). Cette protéine de 70 kDa est très hydrophobe : 95 % des acides aminés sont apolaires (35 % de glycine, 60 % d’alanine, valine, leucine et isoleucine, proline et hydroxyproline). Elle est constituée d’une alternance de séquences hydrophobes et de séquences riches en alanine et en lysine. La tropoélastine est chaperonnée lors du transit intracellulaire par une isoforme de la β-galactosidase (elastin-binding protein). Elle est sécrétée puis déposée sur la charpente de microfibrilles préalablement synthétisée et organisée dans l’espace extracellulaire. Les molécules de tropoélastine s’agrègent entre elles via des interactions hydrophobes avant que les résidus lysine ne soient modifiés par une lysyloxydase [7]. La liaison spontanée de quatre résidus lysine, modifiés ou non, entre les molécules de tropoélastine conduit à la formation des acides aminés de pontage, desmosine et isodesmosine, acides aminés spécifiques de l’élastine. Les molécules de tropoélastine ainsi liées constituent un polymère insoluble dont la fonction principale est de conférer l’élasticité aux tissus (artères, poumon, peau…). L’invalidation du gène unique de l’élastine chez la souris a également démontré le rôle essentiel de l’élastine dans l’artériogenèse : en absence d’élastine, les cellules musculaires lisses artérielles prolifèrent jusqu’à l’occlusion complète des artères, cause du décès des souris, trois à quatre jours après la naissance [8]. Chez l’Homme, l’hémizygotie du gène de l’élastine conduit à la sténose supravalvulaire aortique, pathologie artérielle isolée ou associée au syndrome de Williams-Beuren.
Glycoprotéines de structure
Les glycoprotéines de structure sont des protéines sur lesquelles sont greffées de courtes chaînes glucidiques [9]. Certaines sont ubiquitaires comme la fibronectine, d’autres ont des localisations plus spécifiques comme les laminines des membranes basales. Bien qu’hétérogènes en taille, structure et distribution tissulaire, ces glycoprotéines ont plusieurs points communs : elles contiennent plusieurs domaines structuraux et fonctionnels, plusieurs sites de fixation aux cellules via les intégrines ou autre récepteurs dont le plus fréquent contient la séquence Arg-Gly-Asp (RGD), plusieurs sites d’interactions avec les autres macromolécules extracellulaires. La plupart de ces glycoprotéines sont des multimères.
La fibronectine plasmatique est synthétisée par les hépatocytes et la fibronectine tissulaire par les fibroblastes, les cellules musculaires lisses, les chondrocytes, les cellules endothéliales et épithéliales. Chaque molécule est formée de deux sous-unités de 250 kDa liées par des ponts disulfures. L’existence des différentes isoformes de la fibronectine est due à l’épissage alternatif de l’ARN prémessager. Selon l’origine cellulaire et l’isoforme, les carbohydrates représentent 4 à 10 % de la masse de la molécule. La fibronectine joue un rôle essentiel dans la fixation des cellules à la MEC et l’organisation du cytosquelette. Les laminines sont des hétérotrimères de 850 kDa formés de chaînes α, β et γ [10]. Elles participent à la structure des membranes basales dans lesquelles elles interagissent avec le collagène de type IV, l’entactine/nidogène et le perlécane (protéoglycane à héparane sulfate). Les laminines contrôlent l’adhésion, la prolifération, la différentiation et la polarisation des cellules avec lesquelles elles interagissent. Les autres glycoprotéines de structure sont la vitronectine, l’entactine, les ténascines, les thrombospondines, l’ostéonectine (ou SPARC/BM40) et les glycoprotéines associées à l’élastine. Chacune de ces molécules a une localisation et un rôle spécifique : lors de la synthèse des fibres élastiques, les glycoprotéines forment une charpente sur laquelle se déposent les molécules de tropoélastine (Figure 1B).
Même si les macromolécules de la MEC sont regroupées et décrites en quatre grandes familles, les interactions entre elles sont très nombreuses (élastine-glycoprotéines de structure, collagènes-protéoglycanes, protéoglycanes-glycoprotéines de structure, glycoprotéines de structure-collagènes). Ces interactions participent à la mise en place et au maintien de la cohésion tissulaire.
Mécanismes de vieillissement de la MEC
Chez l’adulte, le renouvellement des macromolécules de la matrice extracellulaire est lent : en mesurant la concentration de D-aspartate, il a ainsi été calculé que la demi-vie de l’élastine dans le parenchyme pulmonaire et l’aorte est de 70 ans, en l’absence de toute pathologie [11]. Deux mécanismes interviennent dans le vieillissement des macromolécules de la matrice extracellulaire : l’interaction avec les facteurs environnementaux (glucose, lipides, calcium…) et l’altération des processus de synthèse et dégradation, les facteurs environnementaux pouvant eux-mêmes modifier les processus de synthèse et de dégradation.
Au cours de leur synthèse, pendant le développement et la croissance, les molécules de collagènes d’une part, et les molécules d’élastine d’autre part, sont liées entre elles. Lors de la formation des acides aminés de pontage, la première étape est la désamisation oxydative de résidus lysine (collagènes et élastine) ou d’hydroxylysine (collagènes) catalysée par une lysyl-oxydase. C’est lors de la formation de ces acides aminés de pontage que les molécules acquièrent leurs fonctions de résistance à l’étirement pour les collagènes et d’élasticité pour l’élastine. Elles acquièrent simultanément leurs propriétés de résistance à la protéolyse. Au cours du vieillissement, d’autres liaisons intermoléculaires sont formées par réaction d’un ose avec les fonctions aminées libres. Toutes les protéines de l’organisme peuvent être oxydées et glyquées mais les conséquences de ces altérations sont plus néfastes sur les protéines de la MEC en raison de leur longue durée de vie dans l’organisme. La glycation non enzymatique, par la liaison de sucres réducteurs comme le glucose aux résidus lysine et arginine à l’extérieur des cellules, va former des produits dits d’Amadori [1]. Si des oxydations ultérieures ont lieu, il y a formation d’AGE (advanced-glycosylation end products) dont les archétypes sont la n-carboxyméthyl-lysine et la pentosidine qui s’accumulent avec le vieillissement et, de façon plus précoce, dans des pathologies comme le diabète. Ces AGE sont des liaisons intermoléculaires supplémentaires qui induisent une augmentation de la résistance à l’étirement pour les collagènes et une diminution de l’élasticité pour l’élastine. Leur formation modifie les chaînes latérales des acides aminés dans les protéines. Ces modifications peuvent également résulter de désamidation, isomérisation, racémisation, oxydation… [12]. De telles altérations des acides aminés perturbent les interactions entre macromolécules de la MEC et entre macromolécules de la MEC et cellules, en particulier lorsqu’elles touchent la séquence RGD.
Dans les différents tissus, les collagènes et les fibres élastiques sont synthétisés dans la période périnatale et dans l’enfance. Au-delà de la phase de croissance, leurs synthèses sont très faibles et leurs vitesses de renouvellement très lentes. La synthèse de ces macromolécules peut-être réactivée au niveau des sites de cicatrisation à la suite d’une blessure mécanique ou d’une agression protéolytique. La synthèse des différentes protéines constitutives des fibres élastiques, parfaitement coordonnée dans le temps et dans l’espace lors du développement, est cependant moins efficace lors de ces étapes de réparation tissulaire. La proportion des différents types de collagènes synthétisés peut également être modifiée.
Avec l’âge, les synthèses de fibronectine et laminine sont augmentées [13]. Cela est vrai pour la fibronectine plasmatique comme pour la fibronectine tissulaire, au niveau des ARNm comme de la protéine. Cette synthèse accrue de fibronectine est également observée au cours du syndrome de Werner. Des altérations des protéoglycanes sont également observées au cours du vieillissement : la longueur et la qualité des glycosaminoglycanes sont modifiées. Lorsque des cellules musculaires lisses artérielles évoluent vers un phénotype migratoire, elles synthétisent moins de protéoglycanes à héparane sulfate, inhibiteurs de la prolifération, et plus de longues chaînes de chondroïtine sulfate, portées principalement par le versicane. Ce changement de qualité des glycosaminoglycanes participerait à la rétention accrue des lipoprotéines LDL dans la paroi artérielle [14].
Le catabolisme des macromolécules de la MEC fait intervenir diverses protéases. Certaines (la métalloprotéinase matricielle MMP-2 des cellules musculaires lisses, par exemple) sont synthétisées et sécrétées par les cellules mésenchymateuses et les cellules épithéliales [3]. Mais, de par le contrôle de l’étape d’activation du zymogène et l’inhibition de son activité par les inhibiteurs synthétisés par les mêmes cellules, l’activité de la MMP-2 est très faible et le catabolisme de la MEC, lent.
Au cours du développement de diverses pathologies liées au vieillissement, d’autres enzymes (élastinolytiques, collagénolytiques…) sont détectables dans les tissus lors des phases inflammatoires aiguës ou chroniques. Celles-ci sont synthétisées et sécrétées par les cellules inflammatoires (polymorphonucléaires neutrophiles, macrophages) ou néosynthétisées par les cellules musculaires lisses stimulées par les cytokines pro-inflammatoires (IL-1β, TNF-α…) [15]. Les activateurs du plasminogène tPA ou uPA peuvent participer à la dégradation de la MEC si la concentration tissulaire de plasminogène est augmentée. La plasmine formée dégrade directement (les glycoprotéines de structure, par exemple) ou indirectement (en activant les métalloprotéinases matricielles) divers composants de la MEC.
La protéolyse péricellulaire des glycoprotéines d’adhésion induit l’anoïkis des cellules, c’est-à-dire l’apoptose consécutive à la rupture des communications entre les cellules et leur environnement. Ce processus est en partie responsable de la disparition des cellules musculaires lisses de la paroi anévrismale [16]. La dégradation des macromolécules de la MEC génère la formation de peptides dont les matrikines qui ont des propriétés biologiques propres et modulent la prolifération et la migration cellulaire, la production des protéases et l’apoptose des cellules [17].
Vieillissement de la MEC vasculaire
Les principales altérations observées au cours du vieillissement dans les grandes artères, chez l’homme comme chez les animaux, sont l’augmentation de la lumière artérielle, l’épaississement de l’intima-média, l’augmentation de la rigidité artérielle et le dysfonctionnement des cellules endothéliales [18] (Figure 2). Chez l’homme, l’épaisseur intima-média augmente d’un facteur deux à trois entre 20 ans et 90 ans. L’épaississement de l’intima, plus important que celui de la média, est dû à la migration des cellules musculaires lisses de la média vers l’intima et à la synthèse de matrice extracellulaire par ces cellules. L’épaississement de la média est le résultat des processus d’hypertrophie des cellules musculaires lisses et l’accumulation des collagènes et de la fibronectine, l’accumulation des collagènes étant directement liée à la formation des AGE. Chez le rat, espèce insensible au processus d’athérosclérose, la quantité absolue d’élastine ne varie pas en fonction de l’âge. Toutefois, du fait de l’augmentation des collagènes et autres macromolécules de la MEC, la proportion relative d’élastine diminue [19]. La diminution de la proportion d’élastine, l’augmentation de la proportion des collagènes auxquelles s’ajoute la fixation du calcium à l’élastine participent à l’accroissement de la rigidité artérielle avec l’âge. Cet accroissement de la rigidité artérielle est associé à une augmentation de la pression artérielle systolique, une diminution de la pression artérielle diastolique, c’est-à-dire une augmentation de la pression pulsée, altération qui peut être évaluée en mesurant la vitesse de l’onde du pouls [20]. Ces altérations sont observées en l’absence de tout état pathologique. Le lien entre la glycation non enzymatique des protéines (dont les collagènes) et la rigidité artérielle a été démontré par la diminution de la rigidité artérielle chez les animaux traités avec l’aminoguanidine qui inhibe la formation des AGE ou avec des « casseurs » d’AGE [21]. Ces altérations des grandes artères observées avec le vieillissement font de l’âge un facteur de risque majeur pour la morbidité-mortalité cardiovasculaire. D’autres altérations des macromolécules de la MEC sont observées au cours des pathologies cardiovasculaires : fixation des lipides sur l’élastine au cours de l’athérosclérose [22], dégradation des fibres élastiques au cours des anévrismes [23] (Figure 2B).
Figure 2
La matrice extracellulaire des artères jeunes (A) et ses modifications au cours du vieillissement et des pathologies artérielles (B).

A. Les artères sont organisées en trois couches concentriques, l’intima, la média et l’adventice et contiennent les quatre catégories de macromolécules de la MEC. Ces macromolécules sont les collagènes, les protéoglycanes (PG), l’élastine et les glycoprotéines de structure dont la fibronectine (FN) et les glycoprotéines associées à l’élastine. Les fibres élastiques artérielles sont organisées en lames fenestrées (LE, lame élastique, LEI, lame élastique interne, LEE, lame élastique externe). La MEC est synthétisée par les cellules endothéliales (CE), les cellules musculaires lisses (CML) et les fibroblastes (F). B. Au cours du vieillissement, l’intima et la média s’épaississent, les cellules musculaires lisses s’hypertrophient, la quantité des collagènes augmente (fibrose) ainsi que la quantité de fibronectine. La quantité de calcium (Ca) lié aux fibres élastiques augmente. L’ensemble de ces processus conduit à la rigidification des artères. Lors de la formation des plaques d’athérome, les lipides transportés par les LDL et les macrophages s’accumulent dans l’espace sous-endothélial. La migration des cellules musculaires lisses de la média vers l’intima et la synthèse de MEC par ces cellules conduit à la formation d’une chape fibreuse plus ou moins importante. Les lipides et le calcium se lient à l’élastine qui devient plus susceptible aux protéases élastolytiques. Lors du développement d’un anévrisme, les fibres élastiques sont intensément hydrolysées et les cellules musculaires lisses disparaissent. L’épaisseur de la média diminue et l’artère se dilate ; l’adventice s’épaissit du fait de la synthèse de collagènes par les fibroblastes. Un thrombus est le plus souvent associé à la paroi anévrismale.
Conclusions
La diversité et la complexité des MEC laissent penser qu’elles sont de véritables puzzles où une seule pièce manquante ou défectueuse peut déstabiliser la structure entière et conduire à des défauts graves dans les propriétés biomécaniques et fonctionnelles d’un tissu. L’organisation tridimensionnelle de chacun des composants et celle des composants les uns par rapport aux autres se mettent en place d’une manière optimale lors du développement et de la croissance de l’organisme. Chacun des composants est remplacé lorsqu’il est altéré ou dégradé. Toutefois, l’organisation tridimensionnelle de la molécule néosynthétisée dans la MEC ne sera jamais optimale, d’où l’intérêt de limiter sa dégradation.
Les altérations de la MEC accentuent-elles le vieillissement des cellules, des tissus, de l’organisme ? Sans doute, ni plus ni moins que les altérations des molécules et des structures intracellulaires, la composition et l’organisation tridimensionnelle de l’une conditionnant le phénotype et les fonctions des autres. Seuls les mécanismes d’altérations des molécules diffèrent, les protéines intracellulaires étant moins sensibles aux modifications post-traductionnelles de par leur vitesse de renouvellement plus rapide [24].
Appendices
Références
- 1. Bailey AJ. Molecular mechanisms of ageing in connective tissues. Mech Ageing Dev 2001 ; 122 : 735-55.
- 2. Labat-Robert J, Robert L. Matrix biology in the 21st century. From a static-rheological role to a dynamic-signaling function. Pathol Biol (Paris) 2005 ; 53 : 369-71.
- 3. Jacob MP. Extracellular matrix remodeling and matrix metalloproteinases in the vascular wall during aging and in pathological conditions. Biomed Pharmacother 2003 ; 57 : 195-202.
- 4. Myllyharju J, Kivirikko KI. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet 2004 ; 20 : 33-43.
- 5. Iozzo RV. Matrix proteoglycans : from molecular design to cellular function. Annu Rev Biochem 1998 ; 67 : 609-52.
- 6. Kielty CM, Sherratt MJ, Shuttleworth CA. Elastic fibres. J Cell Sci 2002 ; 115 : 2817-28.
- 7. Kagan HM, Li W. Lysyl oxidase : properties, specificity, and biological roles inside and outside of the cell. J Cell Biochem 2003 ; 88 : 660-72.
- 8. Li DY, Brooke B, Davis EC, et al. Elastin is an essential determinant of arterial morphogenesis. Nature 1998 ; 393 : 276-80.
- 9. Chothia C, Jones EY. The molecular structure of cell adhesion molecules. Annu Rev Biochem 1997 ; 66 : 823-62.
- 10. Hallmann R, Horn N, Selg M, et al. Expression and function of laminins in the embryonic and mature vasculature. Physiol Rev 2005 ; 85 : 979-1000.
- 11. Powell JT, Vine N, Crossman M. On the accumulation of D-aspartate in elastin and other proteins of the ageing aorta. Atherosclerosis 1992 ; 97 : 201-8.
- 12. Cloos PA, Christgau S. Non-enzymatic covalent modifications of proteins : mechanisms, physiological consequences and clinical applications. Matrix Biol 2002 ; 21 : 39-52.
- 13. Labat-Robert J. Age-dependent remodeling of connective tissue : role of fibronectin and laminin. Pathol Biol (Paris) 2003 ; 51 : 563-8.
- 14. Khalil MF, Wagner WD, Goldberg IJ. Molecular interactions leading to lipoprotein retention and the initiation of atherosclerosis. Arterioscler Thromb Vasc Biol 2004 ; 24 : 2211-8.
- 15. Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis : the good, the bad, and the ugly. Circ Res 2002 ; 90 : 251-62.
- 16. Michel JB. Anoikis in the cardiovascular system : known and unknown extracellular mediators. Arterioscler Thromb Vasc Biol 2003 ; 23 : 2146-54.
- 17. Maquart FX, Bellon G, Pasco S, Monboisse JC. Matrikines in the regulation of extracellular matrix degradation. Biochimie 2005 ; 87 : 353-60.
- 18. Lakatta EG, Levy D. Arterial and cardiac aging : major shareholders in cardiovascular disease enterprises : Part I : aging arteries : a « set up » for vascular disease. Circulation 2003 ; 107 : 139-46.
- 19. Michel JB, Heudes D, Michel O, et al. Effect of chronic ANG I-converting enzyme inhibition on aging processes. II. Large arteries. Am J Physiol 1994 ; 267 : R124-35.
- 20. Laurent S, Boutouyrie P, Lacolley P. Structural and genetic bases of arterial stiffness. Hypertension 2005 ; 45 : 1050-5.
- 21. Corman B, Duriez M, Poitevin P, et al. Aminoguanidine prevents age-related arterial stiffening and cardiac hypertrophy. Proc Natl Acad Sci USA 1998 ; 95 : 1301-6.
- 22. Jacob MP, Hornebeck W, Robert L. Studies on the interaction of cholesterol with soluble and insoluble elastins. Int J Biol Macromol 1983 ; 5 : 275-8.
- 23. Michel JB. Contrasting outcomes of atheroma evolution : intimal accumulation versus medial destruction. Arterioscler Thromb Vasc Biol 2001 ; 21 : 1389-92.
- 24. Gafni A. Protein structure and turnover. In : Masoro EJ, Austad SN, eds. Handbook of the biology of aging. San Diego : Academic Press, 2001 : 59-83.
List of figures
Figure 1
Synthèse des collagènes fibrillaires (A) et des fibres élastiques (B).
A. Chaque triple hélice de collagène résulte de l’association de trois chaînes α codées par un gène (homotrimère) ou plusieurs gènes (hétérotrimère). Les gènes COL1A1, 2, codent pour les chaînes α1 et α2 du collagène I, les gènes COL5A1, 2, 3, 4 pour celles du collagène V et le gène COL3A1 pour celles du collagène III. La triple hélice flanquée de ses extrémités globulaires amino (gros cercles pleins) et carboxyterminale (petits cercles pleins) est sécrétée dans l’espace extracellulaire. Après l’action des amino et carboxyprotéinases, les triples hélices s’associent pour former les fibrilles. Les triples hélices sont liées de manière covalente les unes aux autres lors de la formation des acides aminés de pontage, formation dont la première étape est catalysée par une lysyl-oxidase (LOX ou LOXL). B. Au cours du développement, les microfibrilles formées des glycoprotéines de structure comme les fibrillines et de MAGP (microfibrillar-associated glycoproteins) sont synthétisées et sécrétées dans l’espace extracellulaire ; elles constituent une charpente sur laquelle se déposent les molécules de tropoélastine. Une lysyl-oxydase (LOX ou LOXL) catalyse la première étape de formation des acides aminés de pontage qui résultent de la liaison de deux, trois ou quatre résidus lysine. Les acides aminés de pontage desmosine et isodesmosine sont spécifiques de l’élastine. MFAP2, 5, gène de la MAGP1, 2 ; FBN1, 2 : gène de la fibrilline 1, 2 ; ELN : gène de l’élastine ; GLB1 : gène de la β-galactosidase 1 dont un des ARNm code l’elastin-binding protein.
Figure 2
La matrice extracellulaire des artères jeunes (A) et ses modifications au cours du vieillissement et des pathologies artérielles (B).
A. Les artères sont organisées en trois couches concentriques, l’intima, la média et l’adventice et contiennent les quatre catégories de macromolécules de la MEC. Ces macromolécules sont les collagènes, les protéoglycanes (PG), l’élastine et les glycoprotéines de structure dont la fibronectine (FN) et les glycoprotéines associées à l’élastine. Les fibres élastiques artérielles sont organisées en lames fenestrées (LE, lame élastique, LEI, lame élastique interne, LEE, lame élastique externe). La MEC est synthétisée par les cellules endothéliales (CE), les cellules musculaires lisses (CML) et les fibroblastes (F). B. Au cours du vieillissement, l’intima et la média s’épaississent, les cellules musculaires lisses s’hypertrophient, la quantité des collagènes augmente (fibrose) ainsi que la quantité de fibronectine. La quantité de calcium (Ca) lié aux fibres élastiques augmente. L’ensemble de ces processus conduit à la rigidification des artères. Lors de la formation des plaques d’athérome, les lipides transportés par les LDL et les macrophages s’accumulent dans l’espace sous-endothélial. La migration des cellules musculaires lisses de la média vers l’intima et la synthèse de MEC par ces cellules conduit à la formation d’une chape fibreuse plus ou moins importante. Les lipides et le calcium se lient à l’élastine qui devient plus susceptible aux protéases élastolytiques. Lors du développement d’un anévrisme, les fibres élastiques sont intensément hydrolysées et les cellules musculaires lisses disparaissent. L’épaisseur de la média diminue et l’artère se dilate ; l’adventice s’épaissit du fait de la synthèse de collagènes par les fibroblastes. Un thrombus est le plus souvent associé à la paroi anévrismale.