Article body
La maladie de Huntington (MH) est une maladie neurodégénérative caractérisée par l’apparition progressive de mouvements involontaires de type choréique, des troubles de l’équilibre et des désordres psychiatriques qui apparaissent progressivement. L’issue en est fatale entre 15 et 20 ans après l’apparition des premiers symptômes. Cette maladie héréditaire dominante autosomique est due à la présence anormale de répétitions CAG, codant la glutamine (polyQ), au niveau du gène IT15 qui code la huntingtine [1]. La pathogénie de la MH semble liée à la fois à une perte de fonction de la huntingtine normale et à une toxicité liée à la mutation. Si le rôle précis de la huntingtine normale demeure mal connu - elle a été impliquée dans de multiples fonctions dont la régulation de la transcription, le trafic cellulaire, l’assemblage protéique - elle joue un rôle important dans la neurogenèse au cours du développement. La mutation (polyQ-Htt) induisant des changements de conformation, la huntingtine peut alors être clivée par des caspases, ce qui donne naissance à des fragments amino-terminaux qui s’accumulent pour former des agrégats intraneuronaux insolubles dans les neurites, le cytoplasme et le noyau des cellules. Certains auteurs pensent que ces agrégats sont toxiques parce qu’ils bloquent des fonctions cellulaires vitales : transport axonal, régulation de la transcription de gènes de survie [2-4]. D’autres suggèrent qu’ils sont neuroprotecteurs, et constituent une forme de protection de la cellule contre la toxicité de la forme soluble de polyQ-Htt [5]. Quoi qu’il en soit, ces agrégats représentent la « signature » d’une souffrance neuronale.
Il n’existe aucun traitement de la MH à l’heure actuelle. Une voie d’investigation thérapeutique peut être développée sur la base d’une observation neuropathologique importante, à savoir la vulnérabilité particulière d’une région cérébrale donnée : le noyau caudé (le putamen et le noyau caudé forment une unité fonctionnelle qui correspond au striatum) [6]. Ainsi, bien que la polyQ-Htt soit exprimée de façon ubiquitaire dans le cerveau, seuls les neurones striataux (et parmi ceux-ci, une catégorie de neurones dits « épineux ») dégénèrent dans la MH, tout au moins aux stades les plus précoces de la maladie. Nous avons émis l’hypothèse selon laquelle une sensibilité accrue du noyau caudé dans la MH pouvait être liée aux propriétés neuro-anatomiques de cette région cérébrale, à savoir une dense innervation dopaminergique (DA) issue du mésencéphale. La DA est un neuromodulateur important pour de nombreuses fonctions physiologiques, telles que le contrôle de la coordination motrice, de l’humeur, et les processus d’apprentissage liés à la récompense. Toutefois, sous certaines conditions, libérée ou administrée à forte concentration, la DA peut avoir un effet neurotoxique, in vitro ou in vivo [7]. C’est le cas, par exemple, lorsque de trop fortes doses d’amphétamine (un agoniste indirect de la DA) sont administrées chez l’animal. Des souris génétiquement modifiées qui n’expriment plus le transporteur de la DA (DAT), une protéine qui régule les taux de DA dans le striatum, présentent une dégénérescence spontanée des cellules striatales au cours du vieillissement [8]. De plus, il existe une corrélation directe, chez l’homme, entre la sévérité et l’évolution des symptômes et le gradient dorso-latéral de la concentration en DA [9]. Quelques-uns des effets neurotoxiques de la DA ont été étudiés en relation avec les altérations du métabolisme de l’oxygène et l’on sait que la DA peut s’auto-oxyder spontanément in vitro, ou sous l’influence d’une réaction catalysée par des enzymes in vivo, formant ainsi des radicaux libres. De cette façon, la DA à forte dose (500 µm) permet l’activation d’une voie de signalisation intracellulaire pro-apoptotique, la voie des JNK (cJun N-terminal kinase) qui s’avère déterminante pour induire la mort des neurones striataux [10].
Nous avons pu reproduire deux caractéristiques importantes de la MH : (1) mort progressive des neurones striataux induite par la polyQ-Htt ; (2) formation des agrégats dans les différents compartiments cellulaires (Figure 1), à l’aide d’un modèle in vitro de culture primaire de neurones striataux de souris, dans lesquels des ADN complémentaires (ADNc) codant des formes mutées (polyQ-Htt) ou normale (Htt) de Htt sont introduits par des méthodes de transfection. Il s’agissait donc d’un modèle de choix pour étudier les événements extracellulaires et/ou intracellulaires à l’origine de la vulnérabilité striatale dans la MH.
Figure 1
Modèle cellulaire de maladie de Huntington (MH).

Des neurones striataux embryonnaires (E14) de souris sont mis en culture pendant 7 jours, puis transfectés à l’aide d’ADN complémentaires (ADNc) correspondant à l’exon 1 de la huntingtine normale (Htt) (A) ou mutée (polyQ-Htt) (B). Les protéines traduites fluorescentes sont visualisées en vert, car elles expriment la GFP (green fluorescent protein) en fusion avec les fragments de Htt. Ce marquage nous permet de suivre, au cours du temps, la formation d’agrégats par la huntingtine mutée : dans les neurites, le soma, et le noyau. Des contre-colorations ont été réalisées de façon à visualiser le noyau (marquage au « Hoechst » en bleu), le soma et les neurites (marquage immunocytologique de la protéine MAP2 en rouge) qui nous permettent de voir l’intégrité de la cellule.
Pour étudier le rôle de la DA dans la vulnérabilité des neurones striataux, nous avons combiné des applications de DA, à des doses sub-létales, c’est-à-dire qui induisent peu d’effet sur la mort striatale, à la surexpression des polyQ-Htt. Ce traitement combiné produit une forte amplification des effets toxiques de la polyQ-Htt et accélère à la fois la formation des agrégats et la mort des neurones. Deux événements cellulaires importants sont responsables de ces effets DA (Figure 2).
Figure 2
La dopamine facilite deux événements intracellulaires produits par la huntingtine mutée (polyQ-Htt).
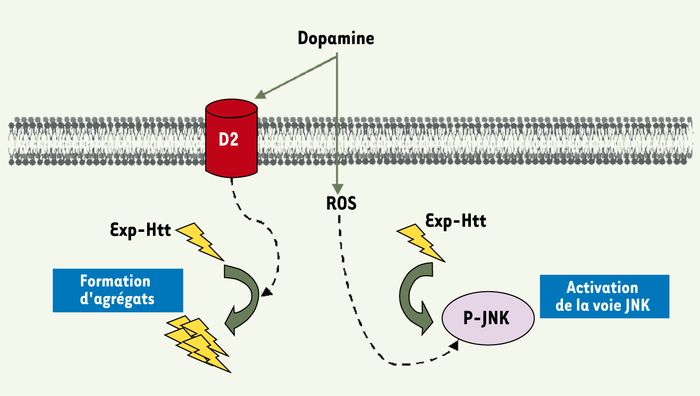
Sous sa forme mutée la huntingtine (polyQ-Htt) est clivée et les fragments ainsi libérés (éclairs jaunes) peuvent s’agréger ou activer la voie JNK dans les cellules. La vulnérabilité des neurones striataux pourrait s’expliquer par un double effet toxique de la dopamine : (1) un effet accélérateur sur la formation des agrégats, via la stimulation des récepteurs D2 ; (2) un effet amplificateur sur la voie JNK, via la production de radicaux libres. Ces deux mécanismes agiraient en synergie pour précipiter la mort des neurones striataux qui expriment la polyQ-Htt.
Tout d’abord, la DA induit un stress oxydatif qui produit, en synergie avec la polyQ-Htt, l’activation de la voie JNK. C’est un mécanisme précoce, apparaissant avant la formation des agrégats nucléaires et avant toute souffrance neuronale. En bloquant soit la production de radicaux libres, soit la voie JNK, une partie significative des effets de la DA sur la mort des neurones striataux est inhibée. Ce mécanisme pourrait être à l’origine d’un accroissement de sensibilité des neurones striataux au cours du vieillissement puisque les systèmes de détoxification des radicaux libres s’amenuisent au cours du temps. À cet égard, il est intéressant de noter que les symptômes, dans la MH, apparaissent chez le jeune adulte, bien que la protéine mutée soit exprimée dès la naissance.
Le deuxième événement est une plus grande quantité d’agrégats polyQ-Htt formés en présence de DA. Cet événement, tout à fait inattendu, implique la stimulation des récepteurs D2. Bloqué par des antagonistes sélectifs D2, cet effet est inexistant dans les neurones striataux provenant d’une souris D2-/-, et peut être reproduit par un agoniste des récepteurs D2. Cela est d’autant plus intéressant, d’un point de vue physiopathologique, que les agrégats de polyQ-Htt sont tout d’abord observés, tant chez l’homme que chez des modèles animaux, dans les régions cérébrales qui expriment le plus de récepteurs D2, dont le globus pallidus, la substance noire compacte, et au sein du striatum, dans les neurones striataux efférents enképhalinergiques [11, 12].
Nos résultats permettent d’expliquer, de façon moléculaire, la vulnérabilité particulière des neurones du striatum dans la MH [13]. Ainsi, l’utilisation conjointe d’un inhibiteur de radicaux libres et d’un antagoniste des récepteurs D2 bloque complètement le rôle stimulateur de la DA sur la mort de ces neurones. Nos résultats apportent également de nouvelles voies d’investigation thérapeutique pour le traitement de la MH, vis-à-vis de laquelle il n’existe aucun traitement à l’heure actuelle. Une thérapie possible serait de donner à des patients diagnostiqués porteurs de la mutation génétique, des inhibiteurs des voies JNK associés à des neuroleptiques, qui sont des antagonistes des récepteurs D2. Notons à cet égard que les patients reçoivent parfois des neuroleptiques, à des stades tardifs, pour traiter les symptômes cognitifs. Nos résultats impliquent des effets précoces de la DA via la stimulation des récepteurs D2. Il serait donc intéressant d’envisager une thérapie à l’aide de cette classe d’agents pharmacologiques à des stades pré-symptomatiques de la maladie.
Appendices
Références
- 1. The Huntington’s disease collaborative research group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993 ; 72 : 971-83.
- 2. Mangiarini L, Sathasivam K, Seller M, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996 ; 87 : 493-506.
- 3. Davies SW, Turmaine M, Cozens BA, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997 ; 90 : 537-48.
- 4. DiFiglia M, Sapp E, Chase KO, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997 ; 277 : 1990-3.
- 5. Arrasate M, Mitra S, Schweitzer ES, et al. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004 ; 431 : 805-10.
- 6. Vonsattel JP, Myers RH, Stevens TJ, et al. Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol 1985 ; 44 : 559-77.
- 7. Jakel RJ, Maragos WF. Neuronal cell death in Huntington’s disease : a potential role for dopamine. Trends Neurosci 2000 ; 23 : 239-45.
- 8. Cyr M, Beaulieu JM, Laakso A, et al. Sustained elevation of extracellular dopamine causes motor dysfunction and selective degeneration of striatal GABAergic neurons. Proc Natl Acad Sci USA2003 ; 100 : 11035-40.
- 9. Aylward EH, Codori AM, Rosenblatt A, et al. Rate of caudate atrophy in presymptomatic and symptomatic stages of Huntington’s disease. Mov Disord 2000 ; 15 : 552-60.
- 10. Luo Y, Umegaki H, Wang X, et al. Dopamine induces apoptosis through an oxidation-involved SAPK/JNK activation pathway. J Biol Chem 1998 ; 273 : 3756-64.
- 11. Reiner A, Albin RL, Anderson KD, et al. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci USA 1988 ; 85 : 5733-7.
- 12. Li H, Li SH, Yu ZX, et al. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington’s disease mice. J Neurosci 2001 ; 21 : 8473-81.
- 13. Charvin D, Vanhoutte P, Pages C, et al. Unraveling a role for dopamine in Huntington’s disease : the dual role of reactive oxygen species and D2 receptor stimulation. Proc Natl Acad Sci USA 2005 ; 102 : 12218-23.
List of figures
Figure 1
Modèle cellulaire de maladie de Huntington (MH).
Des neurones striataux embryonnaires (E14) de souris sont mis en culture pendant 7 jours, puis transfectés à l’aide d’ADN complémentaires (ADNc) correspondant à l’exon 1 de la huntingtine normale (Htt) (A) ou mutée (polyQ-Htt) (B). Les protéines traduites fluorescentes sont visualisées en vert, car elles expriment la GFP (green fluorescent protein) en fusion avec les fragments de Htt. Ce marquage nous permet de suivre, au cours du temps, la formation d’agrégats par la huntingtine mutée : dans les neurites, le soma, et le noyau. Des contre-colorations ont été réalisées de façon à visualiser le noyau (marquage au « Hoechst » en bleu), le soma et les neurites (marquage immunocytologique de la protéine MAP2 en rouge) qui nous permettent de voir l’intégrité de la cellule.
Figure 2
La dopamine facilite deux événements intracellulaires produits par la huntingtine mutée (polyQ-Htt).
Sous sa forme mutée la huntingtine (polyQ-Htt) est clivée et les fragments ainsi libérés (éclairs jaunes) peuvent s’agréger ou activer la voie JNK dans les cellules. La vulnérabilité des neurones striataux pourrait s’expliquer par un double effet toxique de la dopamine : (1) un effet accélérateur sur la formation des agrégats, via la stimulation des récepteurs D2 ; (2) un effet amplificateur sur la voie JNK, via la production de radicaux libres. Ces deux mécanismes agiraient en synergie pour précipiter la mort des neurones striataux qui expriment la polyQ-Htt.