Abstracts
Résumé
Le bras court du chromosome 17 (17p) est fréquemment altéré dans les cancers humains, notamment au niveau du gène p53. Cependant, dans certains cancers, des altérations en 17p concernent des régions distales de p53, en l’absence de toute mutation de ce gène. Des analyses de perte d’hétérozygotie et de méthylation des îlots CpG présents dans les promoteurs de gènes ont permis d’identifier en 17p13.3 plusieurs nouveaux gènes suppresseurs de tumeur proches les uns des autres, tels que HIC1 (hypermethylated in cancer 1) et OVCA1 (ovarian cancer gene 1). HIC1 est d’autant plus intéressant que l’extinction de son expression est, jusqu’à présent, préférentiellement due à l’hyperméthylation de son promoteur, et qu’il a été récemment décrit l’existence d’une boucle de régulation entre HIC1 et p53. Toutefois, si l’impact des modifications épigénétiques dans la tumorigenèse n’est plus à démontrer, les mécanismes orientant le choix d’une inactivation génique par des phénomènes épigénétiques ou génétiques restent à déterminer.
Summary
Loss of heterozygosity (LOH) of the short arm of chromosome 17 (17p) is one of the most frequent genetic alterations in human cancers. Most often, allelic losses coincide with p53 mutations at 17p13.1. However, in many types of solid tumors including sporadic breast cancers, ovarian cancers, medulloblastomas and small cell lung carcinomas, frequent LOH or DNA methylation changes occur in a more telomeric region at 17p13.3, in absence of any p53 genetic alterations. These results suggest that one or more tumor suppressor genes located at 17p13.3 could be involved in tumorigenesis. In addition, the 17p13.3 region has also been implicated in the Miller-Dieker syndrome (MDS), a severe form of lissencephaly accompanied by developmental anomalies caused by heterozygous gene deletions. Analyses of deletion mapping and CpG island methylation patterns have resulted in the identification of two tumor suppressor genes at 17p13.3, HIC1 (hypermethylated in cancer 1) and OVCA1 (ovarian cancer gene 1). HIC1 is a tumor suppressor gene that encodes a transcriptional repressor with five Krüppel-like C2H2 zinc finger motifs and a N-terminal BTB/POZ domain. Clues to the tumor suppressor function of HIC1 have come from the study of heterozygous Hic1+/- mice, which develop spontaneous malignant tumors of different types. Generation of double heterozygous knockout mice Hic1+/-p53+/- provides strong evidence that epigenetically silenced genes such as HIC1 can significantly influence tumorigenesis driven by mutations of classic tumor suppressor genes. This functional cooperation between HIC1 and p53 is interesting and recently, its has been demonstrated that HIC1 was involved in a certain feedback regulation for p53 in tumor suppression through the histone deacetylase SIRT1. However, despite the fact that epigenetic oncogenesis is one of the most vibrant areas of biologic research, the determinants between genetic versus epigenetic routes of tumor suppressor gene inactivation remain elusive.
Article body
Pendant de nombreuses années, la cancérogenèse a été considérée comme découlant de modifications au niveau de la séquence de l’ADN, ces mutations augmentant la fonction de certains gènes, dits oncogènes, ou entraînant une perte de fonction d’autres gènes, dits suppresseurs de tumeur. Dans ce dernier cas, l’inactivation peut également être due à des délétions plus ou moins grandes dans l’ADN.
Depuis 1983, de nombreux travaux ont démontré que la tumorigenèse résulte également de changements épigénétiques, définis comme une modification transmissible du profil d’expression des gènes sans altération de la séquence nucléotidique primaire de l’ADN. Il existe trois principaux types de modifications épigénétiques : l’empreinte génomique, se traduisant par le silence d’un allèle transmis spécifiquement par les parents, les modifications post-traductionnelles (acétylation, méthylation, phosphorylation…) des histones du nucléosome, impliquées dans la régulation transcriptionnelle, et la méthylation de l’ADN, qui correspond au transfert d’un groupe méthyl sur la cytosine des îlots CpG présents au niveau des promoteurs des gènes, contribuant ainsi à l’extinction de l’expression des gènes ciblés. Ainsi, les modifications génétiques (mutations, délétions) comme les modifications épigénétiques sont deux mécanismes permettant d’expliquer la tumorigenèse ; cependant, ces deux processus, loin d’être contradictoires, se révèlent complémentaires lors du développement des cancers, depuis les stades précoces jusqu’aux stades tardifs [1].
HIC1 : un nouveau gène en 17 p13.3 impliqué dans la tumorigenèse
Découverte
L’extinction transcriptionnelle de gènes suppresseurs de tumeurs étant souvent associée à une hyperméthylation des îlots CpG présents dans leur promoteur, il était logique de rechercher de nouveaux gènes suppresseurs de tumeurs sur la base de la détection de régions hyperméthylées de façon aberrante. Dans certains cas, cette hyperméthylation est associée à la perte de la région allélique correspondante, ce qui a notamment été démontré pour le bras court du chromosome 17 (17p). Le chromosome 17 est un « point chaud » comportant plusieurs oncogènes et gènes suppresseurs de tumeurs, dont le gène p53, situé en 17p13.1, qui joue un rôle clé dans la coordination de la réponse de la cellule face à diverses conditions de stress (activation d’oncogènes, hypoxie, dommages à l’ADN). C’est le gène le plus fréquemment muté (50 %) dans les cancers humains non héréditaires.
Cependant, dans certains types de cancers, des altérations en 17p concernent des régions distales de p53, en l’absence même de toute mutation de ce gène. Ces données suggèrent l’existence d’un ou plusieurs gènes suppresseurs de tumeurs situés en 17p13.3, dans une région télomérique par rapport à p53. Le clonage d’un îlot de dinucléotides CpG localisé, en 17p13.3, au niveau du marqueur microsatellitaire D17S30, et méthylé de façon aberrante dans des cancers du poumon, du côlon et du rein [2], a permis d’identifier en 1995 un nouveau gène candidat, appelé HIC1 (hypermethylated in cancer 1). De même, l’identification d’une zone de 15 kb englobant les marqueurs microsatellitaires D17S28 et D17S30, et délétée dans près de 80 % des tumeurs ovariennes, a permis d’isoler en 1996 un autre gène suppresseur de tumeur potentiel, nommé OVCA1 (ovarian cancer gene 1) ou DPH2L1 (diphtamide biosynthesis protein 2-like 1) [3]. Ces études ont souligné l’intérêt de cette région 17p13.3, extrêmement riche en gènes et au sein de laquelle au moins deux gènes, HIC1 et OVCA1, distants de moins de 12 kb, sont impliqués dans la cancérogenèse (Figure 1).
Figure 1
Organisation des gènes suppresseurs de tumeurs dans la région 17p13.

Dans certains types de cancers, des délétions concernent des régions télomériques par rapport à p53 qui est situé en 17p13.1, et ce en l’absence de toute modification de ce gène. La zone 17p13.3, englobant les marqueurs microsatellitaires D17S28 et D17S30, est ainsi délétée dans 85 % des tumeurs de l’ovaire. Elle contient les gènes OVCA1 (DHP2L1), OVCA2 et HIC1. Malgré leurs noms voisins dus à leur expression à partir du même locus génique, les protéines OVCA1 et OVCA2 ont des séquences en acides aminés totalement différentes, et des expériences d’invalidation ont montré que le gène le plus crucial pour la tumorigenèse était OVCA1. Les groupes de S. Baylin et de R. Behringer ont démontré clairement que HIC1 [14] et OVCA1 [22] sont bien des gènes suppresseurs de tumeur ; ainsi sur le chromosome 17, au moins 3 gènes suppresseurs de tumeur (p53, HIC1 et OVCA1) se situent dans une région de moins de 8 Mb. Par ailleurs, la région 17p13.3 est également le siège d’anomalies du gène Lis1, corrélées à une lissencéphalie (absence de circonvolutions du cerveau). Des délétions en 17p13.3, incluant Lis1 mais aussi d’autres gènes comme HIC1 et OVCA1, conduisent au syndrome de Miller-Dieker (forme très sévère de lissencéphalie associée à des malformations), suggérant qu’il existe dans cette région des gènes cruciaux autres que Lis1 impliqués dans le développement.
Le gène HIC1 semble d’autant plus important que les travaux réalisés ces dernières années ont établi qu’il s’agissait d’un suppresseur de tumeurs jouant un rôle de répresseur transcriptionnel, et qu’une boucle de régulation entre les protéines HIC1 et p53 a très récemment été découverte.
Organisation fonctionnelle de la protéine HIC1
Au niveau moléculaire, HIC1 code une protéine de 714 acides aminés, subdivisée en trois régions principales (Figure 2). Sa région aminoterminale comporte un domaine nommé BTB/POZ (broad complex, tramtrack and bric à brac/poxvirus and zinc finger), permettant des interactions protéine/protéine et pouvant réprimer la transcription de façon autonome. Sa région carboxyterminale contient, quant à elle, cinq doigts de zinc de type Krüppel C2H2, fixant de manière coopérative une séquence spécifique de type GGCA au niveau des promoteurs des gènes cibles de HIC1 [4], et une zone conservée dans différentes espèces, mais dont la fonction reste encore inconnue. Enfin, la partie centrale est globalement peu conservée au cours de l’évolution, à l’exception de deux motifs peptidiques correspondant à un site de recrutement du corépresseur CtBP (C-terminal binding protein) de type GLDLSKK [5] et à un site de sumoylation, dont la fonction biologique est jusqu’ici mal connue.
Figure 2
Structure de la protéine HIC1 et implication dans la cancérogenèse.
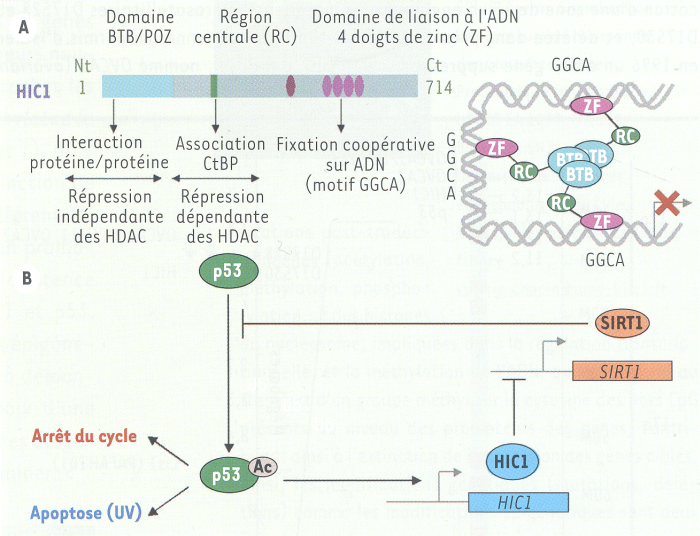
A. HIC1 est un répresseur transcriptionnel contenant un domaine BTB/POZ, une région centrale (RC) et un domaine comportant cinq doigts de zinc (ZF). Dans la région aminoterminale, le domaine BTB/POZ permet des interactions protéine/protéine, d’où la possibilité pour HIC1 de s’oligomériser et de recruter un(des) partenaire(s) impliqué(s) dans les mécanismes de répression. La répression de la transcription induite par le BTB/POZ de HIC1 ne semble pas impliquer d’activité de désacétylation des histones (HDAC) de classe I ou II sensibles à la trichostatine [7, 8]. La région centrale de HIC1 comporte un site de recrutement du corépresseur CtBP, qui réprime la transcription de manière dépendante ou indépendante des HDAC. Dans la région carboxyterminale, seuls quatre des cinq doigts de zinc sont impliqués dans la fixation d’HIC1 sur un motif d’ADN spécifique (GGCA) contenu dans les promoteurs de ses gènes cibles. HIC1 se multimérise pour se fixer de manière coopérative sur l’ADN et réprimer efficacement la transcription de ses gènes cibles [4]. B. Une boucle de régulation entre HIC1 et p53 a été récemment décrite : p53 active la transcription de HIC1, tandis que HIC1 inhibe la transcription de la sirtuine 1 (SIRT1) [27], une HDAC de classe III désacétylant notamment p53, entraînant une diminution de son activité transcriptionnelle. Ainsi, quand HIC1 est absent, SIRT1 est augmentée, ce qui induit une désacétylation de p53 et permettrait notamment aux cellules d’échapper à l’apoptose induite par p53 après une irradiation, augmentant ainsi les risques de cancérisation. BTB/POZ : broad complex, tramtrack and bric à brac/poxvirus and zinc finger ; CtBP : C-terminal binding protein.
De nombreuses protéines à domaine BTB/POZ sont des répresseurs transcriptionnels interagissant classiquement avec des complexes de désacétylation des histones (HDAC, histone deacetylase), ce qui empêcherait le processus de transcription soit en favorisant le passage de la chromatine à un état condensé, soit en inactivant des facteurs de transcription spécifiques ou des facteurs de la machinerie transcriptionnelle de base [6]. Contrairement aux membres de la même famille, la répression de la transcription induite par le domaine BTB/POZ de HIC1 ne semble pas impliquer les complexes multiprotéiques associés à des HDAC de classe I ou II, sensibles à la trichostatine A [7] [8]. Néanmoins, HIC1 est bien associée à une activité HDAC endogène [5] ; par ailleurs, HIC1 interagit avec CtBP, qui peut réprimer la transcription selon deux mécanismes, l’un dépendant, l’autre indépendant du recrutement d’une activité HDAC.
HIC1 est un gène suppresseur de tumeur
Importance des modèles animaux
Depuis la découverte de HIC1 en 1995, l’hyperméthylation de ce gène a été détectée dans de nombreux types de tumeurs, notamment de la prostate [9], de l’estomac [10], de l’ovaire [11] et du sein [12], ainsi que dans des médulloblastomes [13]. Cette méthylation aberrante conduit à une diminution spécifique de l’expression du gène HIC1 dans les cellules tumorales, alors qu’il est exprimé dans les cellules normales correspondantes. Par ailleurs, la réintroduction par transfection stable, dans différents types de lignées cancéreuses, du gène HIC1 sous contrôle d’un promoteur fort aboutit à une inhibition de la prolifération [2]. Toutes ces données indiquent que HIC1 est potentiellement un gène suppresseur de tumeur.
S’il est maintenant admis que l’inhibition de la transcription induite par l’hyperméthylation du promoteur est une cause plutôt qu’une conséquence du processus de tumorigenèse, la question est de savoir si les gènes inactivés du fait de modifications épigénétiques jouent un rôle réel dans l’initiation et la progression tumorale in vivo. Récemment, le rôle de HIC1 en tant que gène suppresseur de tumeur a été clairement démontré grâce à l’étude de souris hétérozygotes Hic1+/- [14] : ces souris développent des tumeurs spontanées avec une incidence beaucoup plus élevée (25 %) que les souris témoins. Les tumeurs apparaissent très tardivement dans la vie de l’animal (70 semaines), ce qui rejoint le cas des souris Pten+/- (phosphatase and tensin homologue) [15]. De manière intéressante, les souris Hic1+/- développent un spectre de tumeurs différant sensiblement selon le sexe de l’animal : les mâles développent de façon prédominante des cancers épithéliaux (75 %) et les femelles des lymphomes et des sarcomes (85 %). De plus, les tumeurs développées par l’ensemble de la population des animaux hétérozygotes sont plutôt de type épithélial (44 %) : ce chiffre est d’autant plus surprenant que ce sont des lymphomes et des tumeurs mésenchymateuses qui sont généralement observés chez les souris âgées ou chez lesquelles un gène a été inactivé ; cette distribution chez les animaux Hic1+/- se rapproche plus de celle des types de cancers constatés chez l’homme [16].
Enfin, la perte de fonction de HIC1 chez les souris hétérozygotes Hic1+/- semble être associée, dans les tumeurs, à une hyperméthylation très importante du promoteur de l’allèle sauvage restant, à l’instar de ce qui est observé chez l’homme [14] ; or c’est normalement la délétion allélique qui est le mécanisme prévalent d’inactivation génique dans le modèle murin, et non l’hyperméthylation. Lors de la cancérisation, la perte d’expression d’un gène peut être due à des pertes d’hétérozygotie, à des mutations ou, encore, à des modifications épigénétiques ; cependant, les mécanismes orientant le choix d’une inactivation préférentiellement par des phénomènes épigénétiques ou par des mutations sont encore mal connus. Dans le cas de HIC1, l’extinction génique semble essentiellement due à l’hyperméthylation, puisque les travaux tentant de découvrir des mutations de ce gène se sont révélés négatifs (Tableau I).
Tableau I
Gènes suppresseurs de tumeur situés dans la région 17p13 (région 13 du bras court du chromosome 17).

Le chromosome 17 est un « point chaud » comportant plusieurs gènes suppresseurs de tumeur importants, et au niveau duquel des délétions sont impliquées dans différents types de cancers. Toutefois, la perte d’expression d’un gène peut être également due à des altérations de la séquence de l’ADN ou à des modifications épigénétiques. Les mécanismes qui orientent le choix d’une inactivation préférentiellement par des mutations ou par des phénomènes d’hyperméthylation des CpG du promoteur des gènes sont encore inconnus. Ainsi, certains gènes tels que p53 présentent essentiellement des mutations, et des cas d’inactivation génique due à une hyperméthylation n’ont été décrit que très récemment (+*). D’autres, tels que OVCA1, sont le plus souvent délétés, et parfois mutés ; l’existence de modifications épigénétiques spécifiques au niveau de ce gène n’a pas encore été décrite (nd), même si des expériences ont montré une hyperméthylation de marqueurs génétiques situés sur des locus proches. Enfin, dans le cas de HIC1, les travaux tentant de découvrir des mutations de ce gène se sont révélés négatifs (-), indiquant que la perte de son expression est jusqu’ici préférentiellement due à l’hyperméthylation. Cependant, l’inactivation génique de HIC1 peut être dans certains cas le résultat d’une délétion (+ ?) : en effet, lors de la délétion de la région englobant les marqueurs D17S28-D17S30 (85 % des cancers de l’ovaire), la perte d’éléments de régulation de HIC1 est une hypothèse envisageable ; par ailleurs, chez les souris hétérozygotes Hic1+/- p53+/-, les tumeurs présentent des délétions entraînant la perte de l’allèle Hic1 sauvage restant [23]. HIC1 : hypermethylated in cancer 1, ; OVCA1 : ovarian cancer gene 1 ; DPH2 : diphtamide biosynthesis protein ; EF-2 : elongation factor 2.
L’ensemble de ces expériences démontre donc que HIC1 appartiendrait à une classe de gènes suppresseurs de tumeur dont l’expression serait éteinte principalement par des modifications épigénétiques, et dont la perte d’une seule copie serait suffisante pour permettre la progression tumorale [17].
D’autres rôles pour HIC1 ?
HIC1 est également un gène candidat pour un syndrome des gènes contigus, le syndrome de Miller-Dieker (MDS). Le MDS appartient à la famille des lissencéphalies, caractérisées par des anomalies des circonvolutions du cerveau et de l’organisation des couches du cortex. La lissencéphalie peut être notamment due à des mutations du gène Lis1 (également nommé PAFAH1B1), localisé en 17p13.3 (Figure 1) et dont la protéine, associée aux microtubules, joue un rôle clé dans la migration des neurones [18]. Le MDS est caractérisé par une forme très sévère de lissencéphalie, accompagnée de dysmorphies faciales et, parfois, d’autres malformations congénitales. Ce syndrome est du à des microdélétions en 17p13.3, incluant le gène Lis1 mais aussi d’autres gènes en cours d’identification.
L’analyse de 30 patients atteints de MDS, en comparaison de patients atteints de lissencéphalie classique, montre qu’ils possèdent tous une zone minimale de délétion en 17p13.3 incluant Lis1, bien sûr, mais également HIC1 et son proche voisin OVCA1 [19]. Le phénotype des souris Hic1-/- présente d’ailleurs des similarités frappantes avec le phénotype des patients atteints de MDS : les embryons, qui meurent juste avant la naissance, sont de petite taille et, surtout, ont de graves anomalies du développement au niveau du crâne, de la face et des membres, ainsi qu’une hernie de la paroi ventrale (omphalocèle) [20]. Par ailleurs, durant le développement embryonnaire des souris, Hic1 est exprimé dans de nombreux organes porteurs d’anomalies chez les patients atteints de MDS [21]. Les souriceaux Ovca1-/- présentent, quant à eux, des similarités avec les animaux Hic1-/- : ils sont de petite taille, meurent juste avant ou dès la naissance et présentent des anomalies du développement (fente palatine, défaut de formation du tube neural au niveau du mésencéphale, polydactylie ou, encore, immaturité des poumons) [22]. Ces résultats suggèrent que HIC1 et OVCA1 sont impliqués dans la prolifération embryonnaire et dans l’organogenèse : la délétion de l’un ou l’autre de ces gènes, en plus de la délétion du gène Lis1, doit sans aucun doute contribuer au développement d’un phénotype de lissencéphalie plus marqué chez les patients atteints de MDS.
Relations entre p53 et la région 17p13.3 lors de la tumorigenèse
La tumorigenèse implique l’activation d’oncogènes et l’inactivation de plusieurs gènes suppresseurs de tumeur distincts. Dans la région 17p13.3, deux gènes suppresseurs de tumeurs sont présents : HIC1, codant un répresseur transcriptionnel, et OVCA1, qui vient d’être identifié en tant que composant de la voie de biosynthèse de la diphtamide sur EF-2 (elongation factor 2). Néanmoins, il semble important de déterminer si des gènes inactivés de façon épigénétique, tel HIC1, ou d’autres gènes plutôt délétés, tel OVCA1, sont capables d’influencer le développement tumoral, et ce dans un contexte bien déterminé d’altération génétique de suppresseurs de tumeur. Dans ce cadre, des souris double hétérozygotes, p53+/- Ovca1+/- ou p53+/- Hic1+/- ont été obtenues.
In vivo, les souris Ovca1+/ développent des tumeurs variées au bout de 92 semaines, à un taux comparable à celui des souris p53+/-, mais avec un spectre différent ; ce temps d’apparition est raccourci à 52 semaines chez les souris p53+/- Ovca1+/- [22]. Les fibroblastes embryonnaires de souris invalidées pour Ovca1 proliférant faiblement, ce gène a été défini comme un régulateur positif de la croissance ; mais la perte d’OVCA1 pourrait aussi conduire à une activation d’un point de contrôle négatif du cycle, dont un excellent candidat est p53, justement situé à proximité sur le chromosome 17 : cela a été confirmé par le fait que les fibroblastes de souris Ovca1-/- p53-/- retrouvent une croissance normale. OVCA1 est donc bien, malgré son rôle positif sur le cycle cellulaire, un gène suppresseur de tumeur impliqué dans la prolifération, le développement embryonnaire et la tumorigenèse. La restauration d’un phénotype normal dans des cellules Ovca1-/- après inactivation de p53, le fait que BRCA1 a été décrit comme pouvant induire l’expression de OVCA1 dans des cellules cancéreuses de sein, et le développement d’un large spectre de tumeurs (et non pas uniquement des tumeurs ovariennes) par les souris hétérozygotes Ovca1+/- sont autant d’indices qui suggèrent que OVCA1 est un intermédiaire dans la réponse cellulaire aux dommages à l’ADN.
Le gène HIC1 est un gène suppresseur de tumeur jouant un rôle négatif dans la prolifération cellulaire. Chez la souris comme chez l’homme, les gènes Hic1 et p53 sont localisés de manière relativement proche sur le même chromosome (chromosome 11 chez la souris). Des souris double hétérozygotes ont été réalisées, chez lesquelles l’altération de ces deux gènes se trouve soit sur des chromosomes séparés (trans), soit sur le même chromosome (cis) [23] (Figure 3). Dans chacun de ces cas, l’altération de Hic1 conduit à des modifications de l’incidence, de la virulence et du spectre des tumeurs par rapport aux souris p53+/-. Les souris double hétérozygotes en cis présentent une apparition plus précoce, ainsi qu’une prévalence et une agressivité accrues des cancers de l’ovaire et des ostéosarcomes, avec une délétion des deux allèles sauvages restants. En revanche, les souris double hétérozygotes en trans ne présentent pas les mêmes mécanismes d’inactivation génique des allèles sauvages restants : la copie sauvage de Hic1 y est souvent hyperméthylée, tandis que la copie sauvage de p53 se trouvant sur le chromosome opposé est préférentiellement délétée (Figure 3). Ainsi, HIC1 et p53 peuvent influencer la tumorigenèse de manière individuelle ou synergique, chacun utilisant pour leur inactivation des mécanismes distincts.
Figure 3
Inactivations épigénétique et génétique de gènes suppresseurs de tumeur lors de la tumorigenèse chez la souris.
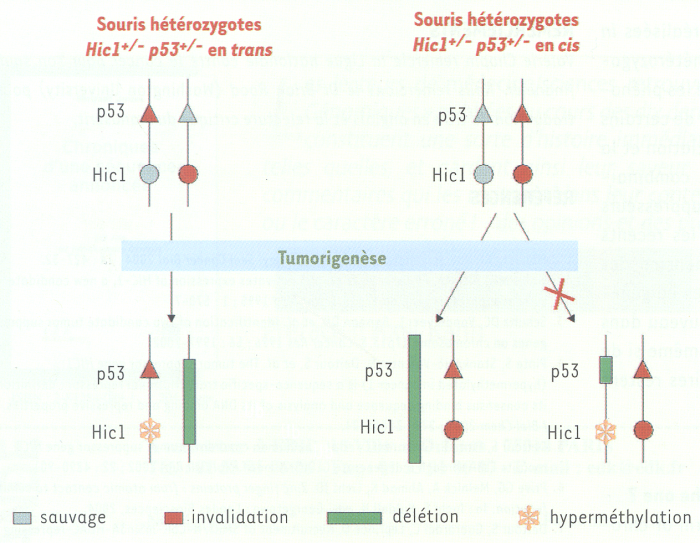
Les gènes Hic1 et p53 sont, chez la souris, tous les deux situés sur le chromosome 11. La création de souris double hétérozygotes Hic1+/- p53+/- a permis de prouver que, lors de la cancérisation, l’inactivation génique de Hic1 et de p53 se produit selon des mécanismes distincts : Hic1 est préférentiellement hyperméthylé, tandis que p53 est le plus souvent délété [23]. En effet, chez les souris trans, l’invalidation de Hic1 (cercle rouge) et de p53 (triangle rouge) se situe sur des chromosomes opposés, et les tumeurs formées comportent en plus de ces mutations de la lignée germinale des délétions de l’allèle sauvage de p53 (rectangle) et une hyperméthylation de l’allèle sauvage de Hic1 (astérisque). Chez les souris cis, l’invalidation de Hic1 et de p53 se situe sur le même chromosome. L’inactivation des deux allèles sauvages restants peut se faire soit par délétion de p53 et hyperméthylation de Hic1, soit par une délétion totale du chromosome opposé comportant ces deux allèles. C’est cette dernière hypothèse qui est validée par l’analyse des tumeurs ; il est évident que la sélection d’une tumorigenèse rapide est favorisée par l’inactivation simultanée des deux gènes (délétion entière du chromosome ne nécessitant qu’un seul et même événement), plutôt que par l’inactivation successive des deux allèles sauvages restants (délétion de p53 et hyperméthylation de Hic1, nécessitant deux événements distincts).
Cette coopération fonctionnelle entre HIC1 et p53 observée in vivo est d’autant plus intéressante que le promoteur de HIC1 contient des sites consensus de liaison de la protéine p53 [2, 24], et que la transfection de p53in vitro active l’expression de HIC1 endogène dans des cellules cancéreuses [2, 25]. Enfin, il a été très récemment démontré que HIC1 est capable d’inhiber la transcription de SIRT1 (codant la protéine sirtuine 1) par l’intermédiaire d’un complexe SIRT1/HIC1 se fixant sur le promoteur de SIRT1 [26]. SIRT1 est une HDAC de classe III désacétylant également p53, ce qui diminue l’activité transcriptionelle de cette dernière [27]. Ainsi, dans les cellules où HIC1 est inactivé, l’expression de SIRT1 serait augmentée, contribuant à la désacétylation de p53 et permettant aux cellules de résister à l’apoptose induite par les dommages à l’ADN (Figure 2). Néanmoins, les relations exactes entre HIC1 et p53, permettant notamment d’expliquer la synergie d’action observée lors de la cancérisation dans les souris double hétérozygotes Hic1+/- p53+/-, doivent encore être déterminées. Il faut noter que SIRT1 est une HDAC de classe III dépendante du NAD et insensible à la trichostatine A ; de plus, en association avec le répresseur BCL11A (B cell leukemia 11 A protein), SIRT1 inhibe la transcription en désacétylant les histones H3 et H4 du promoteur d’un gène rapporteur [28]. Cela pourrait expliquer que la répression transcriptionnelle induite par le domaine BTB/POZ de HIC1 soit insensible à la trichostatine, qui inhibe les HDAC de classe I et II, mais pas de classe III (comme SIRT1).
Conclusions
Plusieurs gènes suppresseurs de tumeurs tels que p53, HIC1 et OVCA1, dont l’inactivation joue un rôle important lors de la tumorigenèse, sont localisés sur le chromosome 17. HIC1 et OVCA1, distants d’à peine 12 kb, se rapprochent notamment d’une nouvelle classe de gènes suppresseurs de tumeurs au comportement haplo-insuffisant (apparition d’un phénotype délétère dès la perte d’un allèle). Toutefois, dans le cas d’une haplo-insuffisance « stricte », l’allèle sauvage restant s’exprime dans les tumeurs [17] ; la situation est légèrement différente dans le cas de HIC1, puisque l’analyse de son statut dans les tumeurs des souris Hic1+/- et Hic1+/-p53+/- indique que la copie sauvage restante de ce gène est également inactivée, le plus souvent par hyperméthylation des CpG [14, 23]. Les mécanismes sous-jacents guidant le choix de la cellule cancéreuse vers une inactivation épigénétique plutôt que génétique ne sont pour l’instant pas connus [29].
Dans le cas de HIC1, l’hyperméthylation et sa perte d’expression sont observées de manière fréquente dans les tumeurs primaires de cancer du sein. De plus, contrairement aux autres tissus normaux où HIC1 n’est pas ou peu méthylé, la moitié des allèles sont déjà méthylés dans le tissu épithélial mammaire normal. Si la perte de HIC1 contribue fortement à l’apparition de cancers, le sein serait donc un tissu à haut risque de développement tumoral, en raison de l’inactivation de ce gène. En accord avec cette hypothèse, l’expression de HIC1 a été associée à une évolution favorable dans le cancer du sein [30]. De même, HIC1 est hyperméthylé à 100 % dans les hyperplasies bénignes de la prostate (considérées comme des situations non cancéreuses), et à 95 % dans les cancers de la prostate [9]. Il est troublant de constater que, dans le cas du sein comme de la prostate, l’hyperméthylation de HIC1 est détectée dans des tissus normaux d’organes hautement sensibles à la régulation hormonale. Ces données, de même que le fait que les souris Hic1+/- développent un spectre de tumeurs différent selon le sexe de l’animal, suggèrent l’existence d’une relation jusqu’ici inexpliquée entre le gène HIC1 et les hormones sexuelles.
De manière plus générale, les expériences réalisées in vivo dans le contexte de souris doublement hétérozygotes ont clairement permis de démontrer que les phénomènes d’extinction épigénétique spécifiques de certains gènes tels que HIC1 peuvent influencer l’initiation et la progression tumorale in vivo, seuls ou en combinaison avec la modification d’autres gènes suppresseurs de tumeur classiques tels que p53. Ainsi, les récents progrès concernant une meilleure compréhension des causes et des conséquences des phénomènes épigénétiques ouvrent un champ d’investigation nouveau dans le domaine de lutte contre le cancer, et ce même si de nombreux mécanismes et acteurs moléculaires restent à découvrir.
Appendices
Remerciements
Valérie Chopin remercie la Ligue nationale contre le cancer pour son soutien financier. Nous remercions le Dr Brian Rood (Washington University) pour la traduction du titre en anglais et la relecture critique du manuscrit.
Références
- 1. Feinberg AP. The epigenetics of cancer etiology. Sem Cancer Biol 2004 ; 14 : 427-32.
- 2. Wales MM, Biel MA, El-Deiry W, et al. p53 activates expression of HIC-1, a new candidate tumour suppressor gene on 17p13.3. Nat Med 1995 ; 1 : 570-7.
- 3. Schultz DC, Vanderveer L, Berman DB, et al. Identification of two candidate tumor suppressor genes on chromosome 17p13.3. Cancer Res 1996 ; 56 : 1997-2002.
- 4. Pinte S, Stankovic-Valentin N, Deltour S, et al. The tumor suppressor gene HIC1 (hypermethylated in cancer 1) is a sequence-specific transcriptional repressor : definition of its consensus binding sequence and analysis of its DNA binding and repressive properties. J Biol Chem 2004 ; 279 : 38313-24.
- 5. Deltour S, Pinte S, Guerardel C, et al. The human candidate tumor suppressor gene HIC1 recruits CtBP through a degenerate GLDLSKK motif. Mol Cell Biol 2002 ; 22 : 4890-901.
- 6. Prive GG, Melnick A, Ahmad K, Licht JD. Zinc finger proteins : from atomic contact to cellular function. In : Iuchi S, Kuldell N, eds. Georgetown : Landes Biosciences, 2004.
- 7. Deltour S, Guerardel C, Leprince D. Recruitment of SMRT/N-CoR-mSin3A-HDAC-repressing complexes is not a general mechanism for BTB/POZ transcriptional repressors : the case of HIC-1 and gammaFBP-B. Proc Natl Acad Sci USA 1999 ; 96 : 14831-6.
- 8. Yang XJ, Gregoire S. Class II histone deacetylases : from sequence to function, regulation, and clinical implication. Mol Cell Biol 2005 ; 25 : 2873-84.
- 9. Yamanaka M, Watanabe M, Yamada Y, et al. Altered methylation of multiple genes in carcinogenesis of the prostate. Int J Cancer 2003 ; 106 : 382-7.
- 10. Kanai Y, Ushijima S, Ochiai A, et al. DNA hypermethylation at the D17S5 locus is associated with gastric carcinogenesis. Cancer Lett 1998 ; 122 : 135-41.
- 11. Pieretti M, Powell DE, Gallion HH, et al. Hypermethylation at a chromosome 17 « hot spot » is a common event in ovarian cancer. Hum Pathol 1995 ; 26 : 398-401.
- 12. Fujii H, Biel MA, Zhou W, et al. Methylation of the HIC-1 candidate tumor suppressor gene in human breast cancer. Oncogene 1998 ; 16 : 2159-64.
- 13. Rood BR, Zhang H, Weitman DM, Cogen PH. Hypermethylation of HIC-1 and 17p allelic loss in medulloblastoma. Cancer Res 2002 ; 62 : 3794-7.
- 14. Chen WY, Zeng X, Carter MG, et al. Heterozygous disruption of Hic1 predisposes mice to a gender-dependent spectrum of malignant tumors. Nat Genet 2003 ; 33 : 197-202.
- 15. Kwabi-Addo B, Giri D, Schmidt K, et al. Haploinsufficiency of the Pten tumor suppressor gene promotes prostate cancer progression. Proc Natl Acad Sci USA 2001 ; 98 : 11563-8.
- 16. DePinho RA. The age of cancer. Nature 2000 ; 408 : 248-54.
- 17. Quon KC, Berns A. Haplo-insufficiency? Let me count the ways. Genes Dev 2001 ; 15 : 2917-21.
- 18. Wynshaw-Boris A, Gambello MJ. LIS1 and dynein motor function in neuronal migration and development. Genes Dev 2001 ; 15 : 639-51.
- 19. Cardoso C, Leventer RJ, Ward HL, et al. Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3. Am J Hum Genet 2003 ; 72 : 918-30.
- 20. Carter MG, Johns MA, Zeng X, et al. Mice deficient in the candidate tumor suppressor gene Hic1 exhibit developmental defects of structures affected in the Miller-Dieker syndrome. Hum Mol Genet 2000 ; 9 : 413-9.
- 21. Grimm C, Sporle R, Schmid TE, et al. Isolation and embryonic expression of the novel mouse gene Hic1, the homologue of HIC1, a candidate gene for the Miller-Dieker syndrome. Hum Mol Genet 1999 ; 8 : 697-710.
- 22. Chen CM, Behringer RR. Ovca1 regulates cell proliferation, embryonic development, and tumorigenesis. Genes Dev 2004 ; 18 : 320-32.
- 23. Chen W, Cooper TK, Zahnow CA, et al. Epigenetic and genetic loss of Hic1 function accentuates the role of p53 in tumorigenesis. Cancer Cell 2004 ; 6 : 387-98.
- 24. Britschgi C, Rizzi M, Grob TJ, et al. Identification of the p53 family-responsive element in the promoter region of the tumor suppressor gene hypermethylated in cancer 1. Oncogene 2005 ; november 21 (online).
- 25. Guerardel C, Deltour S, Pinte S, et al. Identification in the human candidate tumor suppressor gene HIC-1 of a new major alternative TATA-less promoter positively regulated by p53. J Biol Chem 2001 ; 276 : 3078-89.
- 26. Chen W, Wamng DH, Yen RC, et al. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53 dependent DNA damage responses. Cell 2005 ; 123 : 437-48.
- 27. Vaziri H, Dessain SK, Ng-Eaton E, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001 ; 107 : 149-59.
- 28. Senawong T, Peterson VJ, Leid M. BCL11A-dependent recruitment of SIRT1 to a promoter template in mammalian cells results in histone deacetylation and transcriptional repression. Arch Biochem Biophys 2005 ; 434 : 316-25.
- 29. Chen WY, Baylin SB. Inactivation of tumor suppressor genes : choice between genetic and epigenetic routes. Cell Cycle 2005 ; 4 : 10-2.
- 30. Nicoll G, Crichton DN, McDowell HE, et al. Expression of the hypermethylated in cancer gene (HIC-1) is associated with good outcome in human breast cancer. Br J Cancer 2001 ; 85 : 1878-82.
List of figures
Figure 1
Organisation des gènes suppresseurs de tumeurs dans la région 17p13.
Dans certains types de cancers, des délétions concernent des régions télomériques par rapport à p53 qui est situé en 17p13.1, et ce en l’absence de toute modification de ce gène. La zone 17p13.3, englobant les marqueurs microsatellitaires D17S28 et D17S30, est ainsi délétée dans 85 % des tumeurs de l’ovaire. Elle contient les gènes OVCA1 (DHP2L1), OVCA2 et HIC1. Malgré leurs noms voisins dus à leur expression à partir du même locus génique, les protéines OVCA1 et OVCA2 ont des séquences en acides aminés totalement différentes, et des expériences d’invalidation ont montré que le gène le plus crucial pour la tumorigenèse était OVCA1. Les groupes de S. Baylin et de R. Behringer ont démontré clairement que HIC1 [14] et OVCA1 [22] sont bien des gènes suppresseurs de tumeur ; ainsi sur le chromosome 17, au moins 3 gènes suppresseurs de tumeur (p53, HIC1 et OVCA1) se situent dans une région de moins de 8 Mb. Par ailleurs, la région 17p13.3 est également le siège d’anomalies du gène Lis1, corrélées à une lissencéphalie (absence de circonvolutions du cerveau). Des délétions en 17p13.3, incluant Lis1 mais aussi d’autres gènes comme HIC1 et OVCA1, conduisent au syndrome de Miller-Dieker (forme très sévère de lissencéphalie associée à des malformations), suggérant qu’il existe dans cette région des gènes cruciaux autres que Lis1 impliqués dans le développement.
Figure 2
Structure de la protéine HIC1 et implication dans la cancérogenèse.
A. HIC1 est un répresseur transcriptionnel contenant un domaine BTB/POZ, une région centrale (RC) et un domaine comportant cinq doigts de zinc (ZF). Dans la région aminoterminale, le domaine BTB/POZ permet des interactions protéine/protéine, d’où la possibilité pour HIC1 de s’oligomériser et de recruter un(des) partenaire(s) impliqué(s) dans les mécanismes de répression. La répression de la transcription induite par le BTB/POZ de HIC1 ne semble pas impliquer d’activité de désacétylation des histones (HDAC) de classe I ou II sensibles à la trichostatine [7, 8]. La région centrale de HIC1 comporte un site de recrutement du corépresseur CtBP, qui réprime la transcription de manière dépendante ou indépendante des HDAC. Dans la région carboxyterminale, seuls quatre des cinq doigts de zinc sont impliqués dans la fixation d’HIC1 sur un motif d’ADN spécifique (GGCA) contenu dans les promoteurs de ses gènes cibles. HIC1 se multimérise pour se fixer de manière coopérative sur l’ADN et réprimer efficacement la transcription de ses gènes cibles [4]. B. Une boucle de régulation entre HIC1 et p53 a été récemment décrite : p53 active la transcription de HIC1, tandis que HIC1 inhibe la transcription de la sirtuine 1 (SIRT1) [27], une HDAC de classe III désacétylant notamment p53, entraînant une diminution de son activité transcriptionnelle. Ainsi, quand HIC1 est absent, SIRT1 est augmentée, ce qui induit une désacétylation de p53 et permettrait notamment aux cellules d’échapper à l’apoptose induite par p53 après une irradiation, augmentant ainsi les risques de cancérisation. BTB/POZ : broad complex, tramtrack and bric à brac/poxvirus and zinc finger ; CtBP : C-terminal binding protein.
Figure 3
Inactivations épigénétique et génétique de gènes suppresseurs de tumeur lors de la tumorigenèse chez la souris.
Les gènes Hic1 et p53 sont, chez la souris, tous les deux situés sur le chromosome 11. La création de souris double hétérozygotes Hic1+/- p53+/- a permis de prouver que, lors de la cancérisation, l’inactivation génique de Hic1 et de p53 se produit selon des mécanismes distincts : Hic1 est préférentiellement hyperméthylé, tandis que p53 est le plus souvent délété [23]. En effet, chez les souris trans, l’invalidation de Hic1 (cercle rouge) et de p53 (triangle rouge) se situe sur des chromosomes opposés, et les tumeurs formées comportent en plus de ces mutations de la lignée germinale des délétions de l’allèle sauvage de p53 (rectangle) et une hyperméthylation de l’allèle sauvage de Hic1 (astérisque). Chez les souris cis, l’invalidation de Hic1 et de p53 se situe sur le même chromosome. L’inactivation des deux allèles sauvages restants peut se faire soit par délétion de p53 et hyperméthylation de Hic1, soit par une délétion totale du chromosome opposé comportant ces deux allèles. C’est cette dernière hypothèse qui est validée par l’analyse des tumeurs ; il est évident que la sélection d’une tumorigenèse rapide est favorisée par l’inactivation simultanée des deux gènes (délétion entière du chromosome ne nécessitant qu’un seul et même événement), plutôt que par l’inactivation successive des deux allèles sauvages restants (délétion de p53 et hyperméthylation de Hic1, nécessitant deux événements distincts).
List of tables
Tableau I
Gènes suppresseurs de tumeur situés dans la région 17p13 (région 13 du bras court du chromosome 17).
Le chromosome 17 est un « point chaud » comportant plusieurs gènes suppresseurs de tumeur importants, et au niveau duquel des délétions sont impliquées dans différents types de cancers. Toutefois, la perte d’expression d’un gène peut être également due à des altérations de la séquence de l’ADN ou à des modifications épigénétiques. Les mécanismes qui orientent le choix d’une inactivation préférentiellement par des mutations ou par des phénomènes d’hyperméthylation des CpG du promoteur des gènes sont encore inconnus. Ainsi, certains gènes tels que p53 présentent essentiellement des mutations, et des cas d’inactivation génique due à une hyperméthylation n’ont été décrit que très récemment (+*). D’autres, tels que OVCA1, sont le plus souvent délétés, et parfois mutés ; l’existence de modifications épigénétiques spécifiques au niveau de ce gène n’a pas encore été décrite (nd), même si des expériences ont montré une hyperméthylation de marqueurs génétiques situés sur des locus proches. Enfin, dans le cas de HIC1, les travaux tentant de découvrir des mutations de ce gène se sont révélés négatifs (-), indiquant que la perte de son expression est jusqu’ici préférentiellement due à l’hyperméthylation. Cependant, l’inactivation génique de HIC1 peut être dans certains cas le résultat d’une délétion (+ ?) : en effet, lors de la délétion de la région englobant les marqueurs D17S28-D17S30 (85 % des cancers de l’ovaire), la perte d’éléments de régulation de HIC1 est une hypothèse envisageable ; par ailleurs, chez les souris hétérozygotes Hic1+/- p53+/-, les tumeurs présentent des délétions entraînant la perte de l’allèle Hic1 sauvage restant [23]. HIC1 : hypermethylated in cancer 1, ; OVCA1 : ovarian cancer gene 1 ; DPH2 : diphtamide biosynthesis protein ; EF-2 : elongation factor 2.