Abstracts
Résumé
Les tendons contiennent une grande proportion de matrice extracellulaire mais peu de cellules et de vaisseaux sanguins. Par conséquent, le processus physiologique menant à la réparation du tendon lésé dure plus longtemps que pour la plupart des autres tissus. Certains traitements empiriques sont actuellement appliqués dans le but d’accélérer le processus de réparation. Ces approches dogmatiques reposent fortement sur la prémisse selon laquelle l’inflammation est néfaste et sur le fait que les anti-inflammatoires non stéroïdiens (AINS) sont à prescrire durant la phase inflammatoire aiguë ou chronique. Cependant, des données récentes démontrent que certains médiateurs de la réponse inflammatoire sont essentiels à l’organisation du processus de réparation tissulaire. Cet article analysera donc la pluralité des rôles des prostaglandines et les effets des AINS lors des différentes phases du processus de réparation.
Summary
Tendons are extracellular matrix rich structures allowing the transmission of forces generated by skeletal muscles to bones in order to produce movements. Some intrinsic characteristics of tendons, namely hypovascularity and hypocellularity, may explain their slow rate of healing. A growing body of evidence suggests that the inflammatory process, essential for pathogen clearance and injury scavenging, may play opposite functions in tendon healing. For instance, inflammation can lead to degradation of intact collagen and to viable cell death, thereby increasing the functional deficit and recovery period. Paradoxically, many cellular and subcellular events occurring during the inflammatory response lead to the release of a plethora of growth factors that trigger the healing phase. Prostaglandins are implicated in the inflammatory process and may also contribute to the primary steps of tendon healing. Prolonged administration of non steroidal anti-inflammatory drugs (NSAIDs) is a common practice following musculoskeletal injuries. However, there is no clear consensus on the effect of NSAIDs on tendon healing. This review presents a contemporary vision of the inflammatory process following tendon injury and examines the roles of the constitutive and inducible COX-derived prostaglandins. The effect of COX inhibitors will be addressed and special attention will be taken to describe COX-independent effects of these pharmacological inhibitors. Together, this review is an attempt to guide readers toward a more conscientious use of NSAIDs following tendon injuries.
Article body
Quel que soit son degré d’organisation, chaque être vivant élabore une réaction inflammatoire plus ou moins complexe en réponse à un traumatisme ou à l’invasion d’un intrus. Cette réponse inflammatoire a probablement évolué en fonction de son efficacité à prolonger la survie des espèces aux prises avec une agression comportant une composante infectieuse.
Il semble que les cellules inflammatoires activées puissent également provoquer des dommages tissulaires non spécifiques qui pourraient retarder le processus de réparation. Dans les tissus peu vascularisés à faible concentration cellulaire, comme les tendons (Figure 1), le processus de guérison peut s’étendre sur plusieurs mois. Certaines théories stipulent donc que la minimisation des dommages inflammatoires pourrait se traduire par une accélération significative du processus de guérison. De manière contradictoire, plusieurs observations cliniques suggèrent que l’inflammation jouerait un rôle important dans la réparation tissulaire. Les effets contraires de la réponse inflammatoire soulèvent ainsi plusieurs questions. La réaction inflammatoire est-elle optimale pour les blessures de nature aseptique ? Quel est le rôle de chacune des populations de cellules inflammatoires au niveau du site lésé ? Le recours aux anti-inflammatoires non stéroïdiens (AINS) est-il judicieux, et à quel moment faut-il les prescrire ? Qu’implique la modulation de la séquence d’infiltration des différentes populations de cellules inflammatoires ? Dans le but d’élucider l’effet controversé des AINS sur la récupération de la fonction tendineuse post-traumatique, divers mécanismes cellulaires et subcellulaires seront abordés, en particulier les activités plurivalentes des prostaglandines, la régulation de la dégradation de la matrice extracellulaire, l’induction de la mort cellulaire par le stress oxydatif, les interactions neutrophiles-macrophages, ainsi que les effets propres à chacun des différents AINS. Un regard critique sur l’utilisation des AINS, à la suite d’une blessure tendineuse, sera présenté.
Figure 1
Homéostasie du tendon d’Achille sain.

La matrice extracellulaire du tendon est composée de 90 % de collagène, majoritairement de type I. Les fibres de collagène (A) sont regroupées en faisceaux primaires (B), entre lesquels reposent des ténocytes (fibroblastes tendineux) quiescents (C), dont le noyau est fusiforme. En raison de son faible métabolisme, peu de vaisseaux sanguins irriguent la partie centrale du tendon. Le paraténon (enveloppe externe du tendon) (D) est constitué de tissu conjonctif lâche parsemé de fibroblastes et de mastocytes. La densité vasculaire du paraténon est cinq à six fois supérieure à celle de la partie centrale du tendon. En réponse aux stress mécaniques physiologiques, les ténocytes synthétisent à la fois des métalloprotéinases (E) et/ou des molécules de procollagène (F).
Blessure et réaction inflammatoire
La réaction inflammatoire vise à combattre l’agent pathogène, à débarrasser le tissu des débris cellulaires et matriciels, à établir un lien avec la réponse immunitaire acquise et amorcer la réparation tissulaire. Les signaux de danger déclenchés par une altération tissulaire (Figure 2A) activent les cellules sentinelles du tendon : macrophages résidents, fibroblastes et mastocytes, qui libéreront une vaste gamme de molécules pro-inflammatoires (Figure 2B). Parmi ces molécules, on retrouve plusieurs agents vasodilatateurs principalement libérés par les mastocytes, en particulier les prostaglandines (PG) qui résultent de la conversion de l’acide arachidonique par les cyclo-oxygénases (COX) constitutive (COX-1) et inductible (COX-2). L’augmentation du débit sanguin local, l’établissement du gradient chimiotactique et l’activation de l’endothélium des vaisseaux sanguins favorisent la transmigration rapide des neutrophiles. Lorsqu’ils sont activés, les neutrophiles roulent, puis adhèrent et infiltrent rapidement le site lésé (Figure 2C, Figure 4A) pour libérer ensuite un arsenal de molécules pro-inflammatoires, parmi lesquelles des chimiokines, des radicaux libres et des protéinases. La libération de radicaux libres de l’oxygène et de l’azote, durant cette phase catabolique, peut entraîner des dommages cellulaires [1], ainsi que l’apoptose des cellules propres au tissu lésé (Figure 2D) [2]. Il est donc probable que le stress oxydatif puisse provoquer l’apoptose des ténocytes (fibroblastes tendineux) [3] et interférer ainsi avec le processus de réparation du tendon. Les neutrophiles seront suivis par les monocytes, qui infiltreront la lésion pour s’y différencier en macrophages et exercer leurs activités phagocytaires et cytotoxiques (Figure 2E). En accord avec le processus évolutif, il semble donc que des dommages non spécifiques aux cellules du tissu sain soient un prix à payer pour minimiser le risque d’infection.
Figure 2
Événements précoces survenant après une blessure tendineuse.
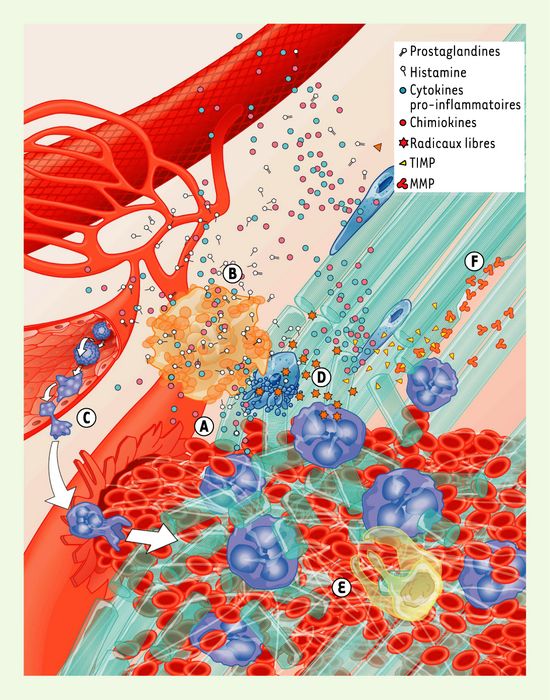
Un traumatisme externe entraîne une désorganisation tissulaire et un saignement (A). Des mastocytes sont activés et libèrent une variété de cytokines pro-inflammatoires, en particulier l’histamine et différentes prostaglandines (B). En conséquence, les neutrophiles roulent, puis adhèrent et infiltrent le site de la lésion (C) où ils libéreront divers médiateurs tels que des radicaux libres, qui endommagent les ténocytes (D). Les macrophages, qui sont en nombre inférieur dans les premiers instants suivant la lésion, contribuent à promouvoir l’environnement pro-inflammatoire, phagocytent les débris et libèrent des molécules cytotoxiques (E). Les métalloprotéinases sécrétées par les cellules inflammatoires dégradent la matrice extracellulaire endommagée sans toutefois détruire les fibres saines. Cette dégradation est réglée par la libération de TIMP (inhibiteurs tissulaires de métalloprotéinases) (F). MMP : métalloprotéinases.
La panoplie de molécules libérées en phase inflammatoire endommagerait également les composantes fonctionnelles des tendons. Ainsi, les enzymes de la famille des métalloprotéinases (MMP) ont la capacité de dégrader différents types de collagène et d’autres composés responsables de la fonction tendineuse. Ces protéinases sont, entre autres, essentielles à la migration des cellules inflammatoires et au remodelage de la matrice extracellulaire. Les MMP produites par les cellules inflammatoires ont la capacité de dégrader le collagène in vitro. Cette action est potentialisée par la présence de radicaux libres qui activent certaines formes latentes de MMP [4]. L’élastase libérée par les neutrophiles augmente également l’activité de plusieurs MMP, dont les MMP-1 et 3, qui ont la capacité de dégrader le collagène. À l’exception des travaux de W. Lee et al. qui établissent un lien entre l’activation des MMP et la dégradation de la matrice extracellulaire [5], peu de preuves ou d’observations directes appuient la théorie des dommages non spécifiques in vivo. Il semble toutefois clair que différents mécanismes puissent réduire les dommages inflammatoires (Figure 2F). Une surexpression rapide des inhibiteurs tissulaires des métalloprotéinases (TIMP) est observée à la suite d’une agression tissulaire aiguë [6]. Ce mécanisme d’autorégulation pourrait être mis en place dans le tissu tendineux. En effet, différents groupes de recherche ont observé une activation des MMP et une expression subséquente ou concomitante de TIMP consécutive à un stress de l’appareil tendineux [7, 8]. Certaines observations suggèrent également que la tension axiale protège les fibres de collagène contre la dégradation protéolytique [9]. Bien que le débat entourant la théorie des dommages inflammatoires ne soit pas clos, des travaux récents montrent que l’accumulation maximale des cellules inflammatoires précède le déficit fonctionnel maximal qui suit un traumatisme tendineux aigu [10]. Ces observations temporelles suggèrent un surpassement des mécanismes de protection intrinsèques des tendons au cours de la phase inflammatoire (Figure 4A).
Résolution de l’inflammation
La résolution de l’inflammation - un processus très contrôlé - est à l’origine de la réparation tissulaire. Après un séjour de 24 à 48 heures, les neutrophiles s’engagent massivement vers la voie de l’apoptose (Figure 3A), en réponse à divers signaux, notamment la diminution des messagers de survie, l’activation des death receptors et l’inhibition du facteur de transcription NF-κB (nuclear factor κB) par les prostaglandines Δ12PGJ2 et 15dPGJ2 [11]. Notons que la prostaglandine D2 (PGD2) et ses métabolites de la famille J pourraient également agir comme anti-inflammatoires via l’activation des récepteurs nucléaires PPAR (peroxisome proliferator-activated receptor). Les neutrophiles apoptotiques sont rapidement phagocytés par les macrophages, donnant à ces derniers un signal qui remplace leur synthèse de cytokines pro-inflammatoires par celle de cytokines anti-inflammatoires. L’ingestion de cellules apoptotiques par les macrophages inhibe la libération de TNF-α (tumor necrosis factor α), d’interleukine-8 (IL-8), de GMCSF (granulocyte-macrophage colony-stimulating factor), d’IL-10, d’IL-1β et de thromboxane A2 [12]. De plus, l’implantation de cellules apoptotiques dans un site inflammatoire stimule la libération du TGF-β1 (transforming growth factor β1) [13]. Le TGF-β1 stimule la résolution de l’inflammation, ainsi que la fibrogenèse. V.A. Fadok et al. [12] ont montré que la prostaglandine PGE2 jouait un rôle majeur dans la commutation des cytokines pro-inflammatoires vers les cytokines anti-inflammatoires dans les macrophages, et que l’administration d’indométacine pouvait inhiber cette commutation, suggérant que l’activité des COX serait importante pour l’amorce du processus de guérison. Des observations récentes indiquent qu’une deuxième vague d’expression de COX-2 en phase de résolution de l’inflammation serait associée à la production de PGD2 [14]. La phagocytose des neutrophiles apoptotiques par les macrophages induirait également un arrêt de la prolifération et un prolongement de la survie de ces derniers [15]. On sait que les macrophages libèrent une panoplie de facteurs de croissance (Figure 3B), dont le TGF-β1, le b-FGF (basic-fibroblast growth factor) et le VEGF (vascular endothelial growth factor), qui sont essentiels pour l’angiogenèse et le démarrage de la réparation tendineuse (Figure 3C-D). Il en découle que la phagocytose des neutrophiles apoptotiques par les macrophages, ainsi que l’activité des COX joueraient des rôles déterminants lors de la résolution de l’inflammation et de la réparation tissulaire. Dans le tendon, tout comme dans la plupart des autres tissus étudiés, les neutrophiles et les macrophages envahissent séquentiellement le site de la blessure, et la présence des macrophages persiste bien au-delà de la disparition des neutrophiles (Figure 4A) [16, 17]. Il devient de plus en plus évident qu’une perturbation de cette séquence physiologique pourrait se traduire par des effets néfastes sur le processus de réparation du tendon (Figure 3E et 4B).
Tableau I
Rôles pro-inflammatoire, anti-inflammatoire et réparateur des prostaglandines.

Figure 3
Résolution de l’inflammation.
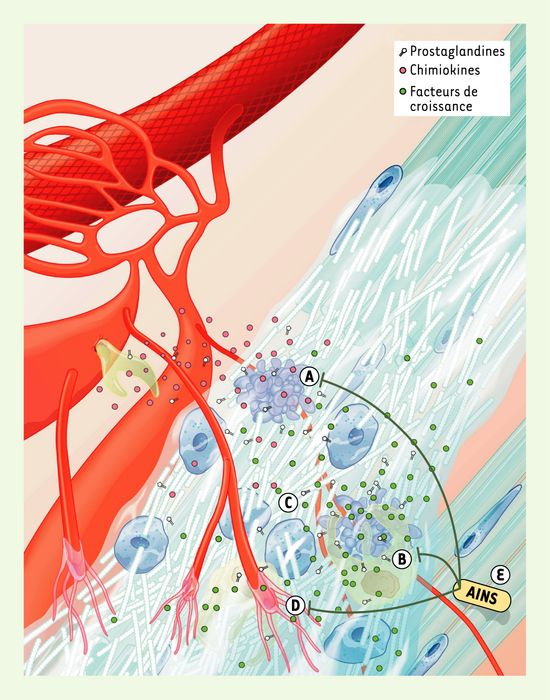
Les signaux de danger ayant disparu, les neutrophiles entrent en apoptose et libèrent des facteurs chimiotactiques pour les macrophages (A). Les macrophages phagocytent les cellules apoptotiques et sécrètent des facteurs stimulant la résolution de l’inflammation, ainsi que le début de la réparation tissulaire (B). En réponse aux facteurs de croissance, les ténocytes prolifèrent et synthétisent une grande quantité de matrice extracellulaire (C) et, simultanément, l’angiogenèse est stimulée (D). Les anti-inflammatoires non stéroïdiens (AINS) pourraient altérer le processus de la réparation tendineuse en modulant l’apoptose, en diminuant la libération de médiateurs anti-inflammatoires et en inhibant l’angiogenèse (E).
Figure 4
Effets hypothétiques de différents traitements par les AINS.

Les neutrophiles et les macrophages infiltrent de façon séquentielle le tendon traumatisé. Durant cette phase précoce, les cellules inflammatoires pourraient accroître le déficit fonctionnel. La résolution de l’inflammation et la réparation tendineuse seront alors mises en marche et le déficit fonctionnel diminuera graduellement (A). L’administration prolongée et continue d’AINS pourrait interférer avec différents processus de la réparation tissulaire et, en fin de compte, ralentir la récupération fonctionnelle (B). L’administration d’AINS en phase pro-inflammatoire limiterait les dommages inflammatoires sans interférer outre mesure avec le processus de réparation tendineuse (C).
AINS et réparation tendineuse
Bien que l’efficacité des AINS dans le traitement des atteintes tendineuses et ligamentaires soit parfois mise en doute, ces médicaments demeurent largement prescrits par le corps médical [18]. Pourtant, certaines études ont révélé que les AINS pouvaient soit améliorer [19], soit détériorer [20], soit n’avoir aucun effet [10] sur les propriétés biomécaniques qui accompagnent un traumatisme tendineux aigu chez l’animal. De nombreux facteurs pourraient expliquer les résultats contradictoires observés, notamment : (1) les effets opposés des diverses prostaglandines qui apparaissent à différentes périodes au cours du processus de réparation ; (2) la spécificité des AINS pour les différentes isoformes des COX ; (3) les modalités d’administration ; (4) les effets secondaires propres à chacun des AINS ; et (5) la région anatomique atteinte. À ce sujet, nous avons observé que le diclofénac diminuait la concentration des cellules inflammatoires uniquement dans la région périphérique du tendon, dont la densité vasculaire est cinq à six fois plus élevée que celle de la partie centrale (Figure 1). Ce traitement n’avait d’ailleurs aucun effet fonctionnel dans un modèle de tendinopathie chez le rat [10]. Une observation relativement constante est que les AINS sont efficaces pour diminuer la sensation de douleur, et ce indépendamment du moment où le traitement démarre. Comme les AINS peuvent influencer une multitude de facteurs clés du processus de réparation, il est encore difficile de statuer de façon définitive sur leur utilité véritable dans le traitement des blessures tendineuses.
Les mécanismes en jeu dans la résolution de l’inflammation seraient mis en route assez tôt, avant la fin de la première semaine qui suit une lésion tendineuse [16, 17]. De manière contradictoire, il n’est pas rare que des AINS soient prescrits pour une période de dix à quinze jours après le traumatisme, et que le début du traitement soit tardif. Ce chevauchement pourrait perturber la libération d’une panoplie de médiateurs anti-inflammatoires et de facteurs de croissance par différents types cellulaires, et en particulier par les macrophages ayant préalablement phagocyté des cellules apoptotiques (Figure 3E). Il est possible que cette perturbation puisse, en fin de compte, retarder la réparation tendineuse (Figure 4B). Dans un tel scénario, l’inhibition de COX-1 par la mutation de son gène retarderait la réparation tissulaire [21] et l’inhibition de COX-2 prolongerait la phase inflammatoire [22]. Des percées récentes en cancérologie [23] ont également permis d’identifier le rôle primordial joué par COX-2 dans le processus d’angiogenèse qui est une condition essentielle à la réparation tissulaire. Ces données sont corroborées par les travaux de B.M. Lapointe et al. [24] qui ont démontré, dans un modèle de blessure musculaire, que la réadaptation à long terme était davantage altérée par un traitement de sept jours par le diclofénac qu’échelonné seulement sur deux jours. De plus amples investigations devront cependant être menées pour clarifier le rôle des COX et des prostaglandines dans le processus de réparation des tissus conjonctifs.
Autres effets des AINS
Certains AINS possèdent des effets physiologiques distincts, sans que ceux-ci ne soient nécessairement liés à l’inhibition des COX. La connaissance de ces mécanismes d’action - c’est-à-dire la connaissance des effets propres à chacune des molécules pharmacologiques - pourrait contribuer à mieux préciser leurs indications respectives. À titre d’exemple, G.P. Riley et al. [25] ont démontré que l’indométacine, ainsi que le naproxène altèrent la prolifération cellulaire et la synthèse de protéoglycanes, tandis que le diclofénac et l’acéclofénac n’affectent pas ces processus. L’utilisation d’indométacine pourrait nuire à la prolifération des ténoblastes mais favoriser la synthèse protéique, suggérant que cet AINS devrait être prohibé durant la phase de prolifération cellulaire mais prescrit au cours de la phase de synthèse protéique [26]. Les AINS bouleversent également une variété d’événements durant la phase inflammatoire mais les corollaires physiologiques de ces perturbations ne sont pas encore compris. Ainsi, le diclofénac induirait la perte de certaines molécules d’adhérence essentielles à l’infiltration des neutrophiles circulants [27] et réduirait la synthèse de leucotriènes par les macrophages [28]. Le diclofénac et l’ibuprofène diminueraient la chimiotaxie des neutrophiles et des macrophages stimulée par le TGF-β1 [29]. De surcroît, certains AINS comme le diclofénac et l’indométacine inhiberaient l’activité d’enzymes capables de dégrader certains médiateurs de l’inflammation comme les lipoxines, tandis que l’ibuprofène, l’acide salicylique, le NS-398, le nimésulide et l’acide niflumique n’auraient pas cette capacité [30]. Ces informations montrent que les AINS devraient être utilisés avec circonspection et que la fenêtre temporelle qui permettrait un effet optimal sur le processus de réparation tendineux n’est pas encore bien définie. Il semble toutefois de plus en plus clair que les traitements prolongés aient plus de chance d’interférer avec la réparation tendineuse que les traitements se limitant à la phase inflammatoire (Figure 4B et 4C).
Conclusions
Plusieurs données scientifiques indiquent que le processus inflammatoire joue un rôle important dans la réparation tissulaire. La modulation de l’inflammation par les AINS fait partie des pratiques médicales depuis plus de cent ans et l’on découvre encore régulièrement de nouvelles vertus à cette classe de composés chimiques. Le manque de données fondamentales et cliniques concernant la perturbation du processus inflammatoire par les AINS fait en sorte qu’une pratique médicale empirique soit encore privilégiée par de nombreux médecins. Il est maintenant reconnu que l’inhibition prolongée des COX puisse provoquer des effets délétères sur le processus de réparation tissulaire et que certains AINS altèrent le métabolisme des ténoblastes. Les AINS seraient donc indiqués dans les tous premiers instants qui suivent le traumatisme afin de réduire les dommages inflammatoires non spécifiques (Figures 4A, 4B). Plusieurs résultats rapportés dans cet article indiquent qu’une utilisation inadéquate des AINS pourrait perturber la réparation tendineuse.
Appendices
Remerciements
Nous tenons à remercier le Dr Claude H. Côté pour sa contribution intellectuelle et M. Gilles Chabot pour les illustrations. Les auteurs ont bénéficié du soutien de différents organismes dont le Fonds de recherche en santé du Québec, le Conseil de recherche en sciences naturelles et génie du Canada, la Fondation canadienne pour l’innovation et la Fondation du Dr Georges Phénix.
Références
- 1. Nguyen HX, Tidball JG. Interactions between neutrophils and macrophages promote macrophage killing of rat muscle cells in vitro. J Physiol 2003 ; 547 : 125-32.
- 2. Galleron S, Borderie D, Ponteziere C, et al. Reactive oxygen species induce apoptosis of synoviocytes in vitro. Alpha-tocopherol provides no protection. Cell Biol Int 1999 ; 23 : 637-42.
- 3. Yuan J, Murrell GA, Trickett A, et al. Involvement of cytochrome c release and caspase-3 activation in the oxidative stress-induced apoptosis in human tendon fibroblasts. Biochim Biophys Acta 2003 ; 1641 : 35-41.
- 4. Maeda H, Okamoto T, Akaike T. Human matrix metalloprotease activation by insults of bacterial infection involving proteases and free radicals. Biol Chem 1998 ; 379 : 193-200.
- 5. Lee W, Aitken S, Sodek J, et al. Evidence of a direct relationship between neutrophil collagenase activity and periodontal tissue destruction in vivo : role of active enzyme in human periodontitis. J Periodontal Res 1995 ; 30 : 23-33.
- 6. Van de Louw A, Jean D, Frisdal E, et al. Neutrophil proteinases in hydrochloric acid- and endotoxin-induced acute lung injury : evaluation of interstitial protease activity by in situ zymography. Lab Invest 2002 ; 82 : 133-45.
- 7. Langberg H, Rosendal L, Kjaer M. Training-induced changes in peritendinous type I collagen turnover determined by microdialysis in humans. J Physiol 2001 ; 534 : 297-302.
- 8. Choi HR, Kondo S, Hirose K, et al. Expression and enzymatic activity of MMP-2 during healing process of the acute supraspinatus tendon tear in rabbits. J Orthop Res 2002 ; 20 : 927-33.
- 9. Nabeshima Y, Grood ES, Sakurai A, et al. Uniaxial tension inhibits tendon collagen degradation by collagenase in vitro. J Orthop Res 1996 ; 14 : 123-30.
- 10. Marsolais D, Côté CH, Frenette J. Nonsteroidal anti-inflammatory drug reduces neutrophil and macrophage accumulation but does not improve tendon regeneration. Lab Invest 2003 ; 83 : 991-9.
- 11. Ward C, Dransfield I, Murray J, et al. Prostaglandin D2 and its metabolites induce caspase-dependent granulocyte apoptosis that is mediated via inhibition of I kappa B alpha degradation using a peroxisome proliferator-activated receptor-gamma-independent mechanism. J Immunol 2002 ; 168 : 6232-43.
- 12. Fadok VA, Bratton DL, Konowal A, et al. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest 1998 ; 101 : 890-8.
- 13. Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF- beta1 secretion and the resolution of inflammation. J Clin Invest 2002 ; 109 : 41-50.
- 14. Gilroy DW, Colville-Nash PR, Willis D, et al. Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med 1999 ; 5 : 698-701.
- 15. Reddy SM, Hsiao KH, Abernethy VE, et al. Phagocytosis of apoptotic cells by macrophages induces novel signaling events leading to cytokine-independent survival and inhibition of proliferation : activation of Akt and inhibition of extracellular signal- regulated kinases 1 and 2. J Immunol 2002 ; 169 : 702-13.
- 16. Marsolais D, Côté CH, Frenette J. Neutrophils and macrophages accumulate sequentially following Achilles tendon injury. J Orthop Res 2001 ; 19 : 1203-9.
- 17. Enwemeka CS. Inflammation, cellularity, and fibrillogenesis in regenerating tendon : implications for tendon rehabilitation. Phys Ther 1989 ; 69 : 816-25.
- 18. Mazzone MF, McCue T. Common conditions of the Achilles tendon. Am Fam Physician 2002 ; 65 : 1805-10.
- 19. Dahners LE, Gilbert JA, Lester GE, et al. The effect of a nonsteroidal antiinflammatory drug on the healing of ligaments. Am J Sports Med 1988 ; 16 : 641-6.
- 20. Elder CL, Dahners LE, Weinhold PS. A cyclooxygenase-2 inhibitor impairs ligament healing in the rat. Am J Sports Med 2001 ; 29 : 801-5.
- 21. Kampfer H, Brautigam L, Geisslinger G, et al. Cyclooxygenase-1-coupled prostaglandin biosynthesis constitutes an essential prerequisite for skin repair. J Invest Dermatol 2003 ; 120 : 880-90.
- 22. Wallace JL, Bak A, McKnight W, et al. Cyclooxygenase 1 contributes to inflammatory responses in rats and mice : implications for gastrointestinal toxicity. Gastroenterology 1998 ; 115 : 101-9.
- 23. Iniguez MA, Rodriguez A, Volpert OV, et al. Cyclooxygenase-2 : a therapeutic target in angiogenesis. Trends Mol Med 2003 ; 9 : 73-8.
- 24. Lapointe BM, Frémont P, Côté CH. Adaptation to lengthening contractions is independent of voluntary muscle recruitment but relies on inflammation. Am J Physiol Regul Integr Comp Physiol 2002 ; 282 : R323-9.
- 25. Riley GP, Cox M, Harrall RL, et al. Inhibition of tendon cell proliferation and matrix glycosaminoglycan synthesis by non-steroidal anti-inflammatory drugs in vitro.J Hand Surg Br 2001 ; 26 : 224-8.
- 26. Almekinders LC, Baynes AJ, Bracey LW. An in vitro investigation into the effects of repetitive motion and nonsteroidal antiinflammatory medication on human tendon fibroblasts. Am J Sports Med 1995 ; 23 : 119-23.
- 27. Diaz-Gonzalez F, Gonzalez-Alvaro I, Campanero MR, et al. Prevention of in vitro neutrophil-endothelial attachment through shedding of L-selectin by nonsteroidal antiinflammatory drugs. J Clin Invest 1995 ; 95 : 1756-65.
- 28. Kothari HV, Lee WH, Ku EC. An alternate mechanism for regulation of leukotriene production in leukocytes : studies with an anti-inflammatory drug, sodium diclofenac. Biochim Biophys Acta 1987 ; 921 : 502-11.
- 29. Locatelli L, Sacerdote P, Mantegazza P, et al. Effect of ibuprofen and diclofenac on the chemotaxis induced by substance P and transforming growth factor-beta on human monocytes and polymorphonuclear cells. Int J Immunopharmacol 1993 ; 15 : 833-8.
- 30. Clish CB, Sun YP, Serhan CN. Identification of dual cyclooxygenase-eicosanoid oxidoreductase inhibitors : NSAIDs that inhibit PG-LX reductase/LTB(4) dehydrogenase. Biochem Biophys Res Commun 2001 ; 288 : 868-74.
List of figures
Figure 1
Homéostasie du tendon d’Achille sain.
La matrice extracellulaire du tendon est composée de 90 % de collagène, majoritairement de type I. Les fibres de collagène (A) sont regroupées en faisceaux primaires (B), entre lesquels reposent des ténocytes (fibroblastes tendineux) quiescents (C), dont le noyau est fusiforme. En raison de son faible métabolisme, peu de vaisseaux sanguins irriguent la partie centrale du tendon. Le paraténon (enveloppe externe du tendon) (D) est constitué de tissu conjonctif lâche parsemé de fibroblastes et de mastocytes. La densité vasculaire du paraténon est cinq à six fois supérieure à celle de la partie centrale du tendon. En réponse aux stress mécaniques physiologiques, les ténocytes synthétisent à la fois des métalloprotéinases (E) et/ou des molécules de procollagène (F).
Figure 2
Événements précoces survenant après une blessure tendineuse.
Un traumatisme externe entraîne une désorganisation tissulaire et un saignement (A). Des mastocytes sont activés et libèrent une variété de cytokines pro-inflammatoires, en particulier l’histamine et différentes prostaglandines (B). En conséquence, les neutrophiles roulent, puis adhèrent et infiltrent le site de la lésion (C) où ils libéreront divers médiateurs tels que des radicaux libres, qui endommagent les ténocytes (D). Les macrophages, qui sont en nombre inférieur dans les premiers instants suivant la lésion, contribuent à promouvoir l’environnement pro-inflammatoire, phagocytent les débris et libèrent des molécules cytotoxiques (E). Les métalloprotéinases sécrétées par les cellules inflammatoires dégradent la matrice extracellulaire endommagée sans toutefois détruire les fibres saines. Cette dégradation est réglée par la libération de TIMP (inhibiteurs tissulaires de métalloprotéinases) (F). MMP : métalloprotéinases.
Figure 3
Résolution de l’inflammation.
Les signaux de danger ayant disparu, les neutrophiles entrent en apoptose et libèrent des facteurs chimiotactiques pour les macrophages (A). Les macrophages phagocytent les cellules apoptotiques et sécrètent des facteurs stimulant la résolution de l’inflammation, ainsi que le début de la réparation tissulaire (B). En réponse aux facteurs de croissance, les ténocytes prolifèrent et synthétisent une grande quantité de matrice extracellulaire (C) et, simultanément, l’angiogenèse est stimulée (D). Les anti-inflammatoires non stéroïdiens (AINS) pourraient altérer le processus de la réparation tendineuse en modulant l’apoptose, en diminuant la libération de médiateurs anti-inflammatoires et en inhibant l’angiogenèse (E).
Figure 4
Effets hypothétiques de différents traitements par les AINS.
Les neutrophiles et les macrophages infiltrent de façon séquentielle le tendon traumatisé. Durant cette phase précoce, les cellules inflammatoires pourraient accroître le déficit fonctionnel. La résolution de l’inflammation et la réparation tendineuse seront alors mises en marche et le déficit fonctionnel diminuera graduellement (A). L’administration prolongée et continue d’AINS pourrait interférer avec différents processus de la réparation tissulaire et, en fin de compte, ralentir la récupération fonctionnelle (B). L’administration d’AINS en phase pro-inflammatoire limiterait les dommages inflammatoires sans interférer outre mesure avec le processus de réparation tendineuse (C).
List of tables
Tableau I
Rôles pro-inflammatoire, anti-inflammatoire et réparateur des prostaglandines.