Abstracts
Résumé
L’étude d’une forme rare d’hypertension artérielle de transmission mendélienne, l’hypertension hyperkaliémique familiale (HHF), a récemment permis d’identifier des mutations dans les gènes WNK1 et WNK4, qui codent pour des protéines appartenant à une nouvelle famille de sérine-thréonine kinases (with no lysine [K] kinase). Plusieurs éléments du tableau clinique de l’HHF, caractérisé par une hyperkaliémie, une hyperchlorémie et une grande sensibilité aux diurétiques thiazidiques, sont en faveur d’une anomalie du transport ionique dans le tubule distal rénal. En accord avec cette hypothèse, WNK1 et WNK4 sont fortement exprimés dans cette partie du néphron. On les retrouve également dans de nombreux épithéliums impliqués dans le transport du chlore, tels que celui du côlon. In vitro, WNK4 règle à la fois le transport de Na+, K+ et Cl-, et pourrait donc constituer une voie de régulation importante des transports ioniques rénal et extra-rénal.
Summary
Arterial hypertension is a complex trait influenced by a variety of environmental and genetic factors. Several approaches can be used to identify its susceptibility genes : one is to study rare monogenic forms of hypertension, like familial hyperkalemic hypertension (FHH). Also known as pseudohypoaldosteronism type 2 or Gordon syndrome, FHH is characterized by hypertension, hyperkalemia despite normal renal glomerular filtration rate, abnormalities which are particularly sensitive to thiazide diuretics. Mild hyperchloremia, metabolic acidosis, and suppressed plasma renin activity are associated findings. Despite its phenotypic and genetic heterogeneity, mutations in two related genes, WNK1 and WNK4, were recently identified. These genes belong to a newly identified family of serine-threonine (with no lysine [K]) kinases. Both are highly expressed in the kidney and in a variety of epithelia involved in chloride transport. It has thus been postulated that these two kinases could be implicated in a new pathway of ionic transport regulation. Several studies have very recently confirmed this hypothesis in vitro, in Xenopus oocytes or kidney cell lines. They have shown that, in the renal distal tubule, WNK4 inhibits sodium reabsorption and potassium secretion, via inhibition of NCC (thiazide-sensitive Na+-Cl- cotransporter) and K+ channel ROMK activity, respectively. Interestingly, FHH mutations have opposite effects : while they lead to loss of NCC inhibition, they increase ROMK inhibition. Moreover, they also increase paracellular permeability to chloride of MDCK cells. WNK4 also inhibits apical and basal chloride transporters present in extra-renal epithelia, such as CFEX and Na+-K+-2 Cl-, respectively. It is also interesting to note that the WNK4-mediated negative regulation of NCC activity is in turn inhibited by WNK1. By its role on several transporters, WNK4 appears as a putative key regulator of ionic transport and blood pressure.
Article body
L’hypertension artérielle (HTA) représente l’exemple même d’une maladie complexe, soumise à des facteurs génétiques et environnementaux divers, dont il est très difficile d’identifier les gènes de susceptibilité. Une des alternatives d’étude est l’identification des gènes impliqués dans certaines formes rares mais caricaturales d’HTA, dont la transmission mendélienne facilite l’analyse génétique. L’hypertension hyperkaliémique familiale (HHF) ou pseudohypoaldostéronisme de type 2 (PHA2) est une forme rare d’hypertension artérielle, autosomique dominante, associant hyperkaliémie, acidose métabolique hyperchlorémique et fonction rénale normale. Ses mécanismes physiopathologiques sont restés longtemps controversés [1]. Cependant, la très grande efficacité des diurétiques thiazidiques chez les sujets atteints et le fait que cette affection se présente comme le miroir du syndrome de Gitelman[1] ont orienté fortement vers une anomalie du tubule distal rénal. Malgré l’hétérogénéité phénotypique et génétique de la maladie [2], nous avons récemment identifié, en collaboration avec l’équipe de R. Lifton (Yale University, New Haven, États-Unis), les gènes correspondant à deux des trois locus préalablement identifiés : WNK1 et WNK4 [3]. Les mutations au locus WNK1 dans les familles américaine et française sont des délétions dans l’intron 1 du gène, de taille respective 41 et 22 kpb, qui ont vraisemblablement pour conséquence la modification de l’expression de WNK1. Dans le cas de WNK4, des mutations faux-sens ont été mises en évidence dans les exons 7 et 17. Elles pourraient modifier l’interaction de WNK4 avec ses partenaires.
Une nouvelle famille de sérine/thréonine kinases
WNK1 et WNK4 appartiennent à une nouvelle famille de sérine/thréonine kinases, la famille WNK (with no lysine [K] kinase), récemment identifiée et peu caractérisée (Figure 1). Ces kinases sont particulières : en effet, la lysine catalytique présente dans le sous-domaine II de presque toutes les protéine kinases connues est remplacée par une cystéine [3, 4]. Outre le domaine kinase, les protéines WNK possèdent un domaine auto-inhibiteur de l’activité kinase [5], deux domaines coiled-coil et trois domaines riches en proline [6]. Malgré leur très forte homologie, les domaines kinase de WNK1 et WNK4 ne semblent pas équivalents. Le domaine kinase de WNK1 est actif in vitro et capable d’autophosphorylation, ce qui n’est pas le cas de celui de WNK4 [5, 7]. Il est possible que l’activation de WNK4 nécessite l’interaction avec un partenaire protéique, comme le suggère l’action du domaine auto-inhibiteur de WNK1 sur WNK4 et l’agrégation de WNK1 in vitro sous forme de tétramère [5, 7]. De même, on ne connaît pas encore toutes les voies activées par les WNK, mais l’équipe de M. Cobb a récemment montré que WNK1 est capable de phosphoryler ERK5 [8].
Figure 1
Localisations chromosomiques, structures géniques et domaines fonctionnels conservés des WNK (with no lysine K kinase).
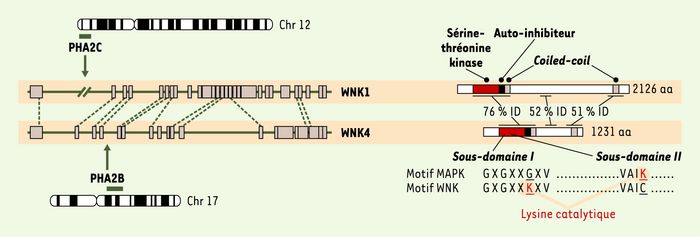
Les gènes correspondant aux locus PHA2B (17p11-q21) et PHA2C (12p13.3) sont des homologues : WNK1 et WNK4 appartiennent à la famille des sérine-thréonine kinases WNK, caractérisée par l’absence de la lysine active dans le sous-domaine II du domaine kinase. La comparaison des séquences nucléiques (partie gauche de la figure) et protéiques (partie droite) met en évidence trois zones d’homologie. Le domaine catalytique sérine-thréonine kinase, le domaine auto-inhibiteur et les deux domaines coiled-coil (hélices hydrophobes) sont conservés.
WNK, néphron distal et transport ionique
Plusieurs données récentes suggèrent que WNK1 et WNK4 constituent une nouvelle voie de régulation du transport ionique rénal et extra-rénal : ces deux kinases sont exprimées dans le tube contourné distal et le canal collecteur [3, 9, 10], mais aussi dans les épithéliums impliqués dans le transport du chlore [11, 12] ; elles sont capables d’autophosphorylation, cette capacité étant influencée par des modifications de la concentration ionique extracellulaire [4] ; elles peuvent régler in vitro plusieurs transporteurs ioniques (voir plus loin).
La portion du néphron distal faisant suite à la macula densa est constitué de trois parties morphologiquement et fonctionnellement différentes : le tubule contourné distal (DCT), le tubule connecteur (CNT) et le canal collecteur cortical (CCD) (Figure 2). Le néphron distal joue un rôle crucial dans l’homéostasie hydrosodée puisque c’est là que sont ajustés de façon fine la quantité de sodium réabsorbé et de potassium sécrété ainsi que le pH urinaire. Cet ajustement est en grande partie dépendant du système rénine-angiotensine-aldostérone. Un défaut de transport ionique dans le néphron distal peut donc avoir des répercussions importantes sur l’équilibre ionique et acido-basique de l’organisme entier. Le transport ionique peut se faire de façon trans- ou paracellulaire. Plusieurs transporteurs ou canaux épithéliaux ont été caractérisés au niveau du néphron distal et leur rôle dans la régulation de la réabsorption sodée ou la sécrétion de potassium a été étudié d’abord in vitro puis, plus récemment, in vivo par inactivation ciblée chez la souris (pour revue, voir [13]). Ceux qui nous intéressent plus particulièrement sont le cotransporteur Na+Cl- apical sensible aux diurétiques thiazidiques (NCC), exprimé dans le DCT, responsable de près de 5 % de la réabsorption sodée rénale (l’expression de NCC est contrôlée en grande partie par les variations de l’apport sodé dans l’alimentation [14]) et le canal potassique apical ROMK, exprimé dans les cellules principales du CCD, site majeur de sécrétion du potassium. L’expression de ROMK est contrôlée par les apports potassiques [15] ou par l’aldostérone via la SKG1 (serum glucocorticoid kinase 1) [16].
Figure 2
Transport du Cl-, du Na+ et du K+ dans le néphron distal et hypothèses physiopathologiques suggérées dans l’hypertension hyperkaliémique familiale (HHF).

Représentation schématique du néphron et des cellules qui composent le tubule contourné distal (DCT) et le canal collecteur cortical (CCD). Ce dernier est constitué de trois types cellulaires : les cellules principales et les cellules intercalaires de types A et B figurées de haut en bas. Les pompes sont représentées en bleu, les échangeurs en orange, les cotransporteurs en violet et les canaux en vert. Le récepteur minéralocorticoïde (MR) est représenté en jaune, les claudines 1-4 en bleu dans le complexe de jonction serrée représenté en rose. À gauche, sont schématisés les mécanismes d’action des WNK suggérés par les expériences réalisées dans les oeufs de xénope et les cellules rénales en culture. À droite, sont schématisées les conséquences des mutations WNK4. Ces différents mécanismes physiopathologiques aboutissent à une augmentation de la réabsorption de Na+ et Cl- et une diminution de la sécrétion de K+ expliquant les anomalies biologiques (hyperkaliémie, hyperchlorémie) et l’hypertension artérielle . CNT : tubule connecteur ; CCM : canal collecteur médullaire ; NCC : cotransporteur Na+Cl- apical sensible aux diurétiques thiazidiques ; ROMK : canal potassique apical.
WNK1 et WNK4 sont exprimés dans de nombreux épithéliums
Des études de profil d’expression par Northern blot ont montré que, bien qu’ubiquitaire, WNK1 est fortement exprimé dans le rein, le cerveau, le muscle squelettique et le coeur. Le profil d’expression de WNK4 est plus restreint mais on retrouve une forte expression dans le rein. Des études d’immunohistochimie [3] ou d’hybridation in situ [9, 10] ont permis de localiser plus précisément l’expression de ces kinases dans le rein : WNK4 est exprimé dans le DCT, le CNT et le CCD. Le cas de WNK1 est plus complexe, puisqu’une de ses isoformes est exprimée faiblement dans l’ensemble du néphron, alors que l’isoforme dite rénale est exprimée uniquement et fortement dans le DCT et le CNT (voir plus loin).
WNK1 et WNK4 sont également co-exprimés dans de nombreux épithéliums impliqués dans le transport du chlore [11, 12] tels que le côlon, les glandes sudoripares, les canaux et la vésicule biliaire ou la peau. Il est intéressant de noter que la localisation cellulaire de WNK1 varie dans ces différents épithéliums : cytoplasmique dans le DCT ou la vésicule biliaire, la protéine est retrouvée au contact des membranes latérales dans le côlon, le pancréas ou les canaux biliaires. De même, il semble que WNK4 soit cytoplasmique dans le CCD et membranaire dans le DCT [3]. L’importance fonctionnelle de cette localisation différentielle reste à élucider.
WNK et réabsorption sodée dans le DCT
Une des caractéristiques des patients atteints de HHF est leur grande sensibilité aux diurétiques thiazidiques qui inhibent l’activité de NCC. Utilisant les oeufs de xénope, deux équipes ont montré que WNK4 inhibe l’activité de transport de NCC, réduisant ainsi la réabsorption de sodium [17, 18] (Figure 2). Cette inhibition semble liée à une réduction du nombre de transporteurs présents dans la membrane et une interaction entre WNK4 et le domaine carboxyterminal de NCC a été mise en évidence par co-immunoprécipitation. F.H. Wilson et al. [17] ont montré que le domaine kinase de WNK4 est nécessaire à cette action. En effet, l’inactivation de l’activité kinase par mutagenèse entraîne un retour à la normale de l’activité de transport de NCC et du nombre de canaux à la membrane. Les quatre mutations faux-sens (Glu562Lys, Asp564Ala, Gln565Glu et Arg1185Cys) actuellement identifiées chez des patients atteints de HHF sont regroupées dans des motifs très conservés de 10 et 16 acides aminés en aval des domaines coiled-coil(Figure 3). L’effet des trois mutations situées dans une séquence très conservée en aval du CC1 (coiled-coil 1) a été testé [18] : seule la mutation Q565E conduit à la perte de l’inhibition exercée par WNK4 sur NCC [17].
Figure 3
Mutations de WNK4 à l’origine de l’hypertension hyperkaliémique familiale (HHF).

La structure exon/intron du gène humain est schématisée en haut de la figure, ainsi que le domaine kinase, le domaine auto-inhibiteur (AI) et les domaines coiled coil (CC1 et CC2). Les quatre mutations faux-sens identifiées dans plusieurs familles atteintes d’HHF sont localisées au niveau de courts motifs conservés de 10 et 16 acides aminés (aa) en aval des domaines coiled coil. Chacune de ces mutations modifie un résidu conservé entre les quatre membres de la famille WNK.
WNK1 est également exprimé dans le DCT. C.L. Yang et al. ont montré, par injection dans l’oeuf de xénope, que l’isoforme longue de WNK1 lève l’inhibition exercée par WNK4 sur NCC [18]. Les mécanismes gouvernant cette action restent à déterminer. Il reste aussi à démontrer que les différentes isoformes de WNK1, avec ou sans activité kinase, ont la même action. En effet, le gène WNK1, d’expression ubiquitaire, est à l’origine de plusieurs transcrits (Figure 4) produits grâce à : (1) un épissage alternatif des exons 9, 11 et 12, séparément ou conjointement ; (2) la présence de deux sites de polyadénylation ; (3) l’existence de plusieurs promoteurs.
Figure 4
Mutations de WNK1 à l’origine de l’hypertension hyperkaliémique familiale (HHF).
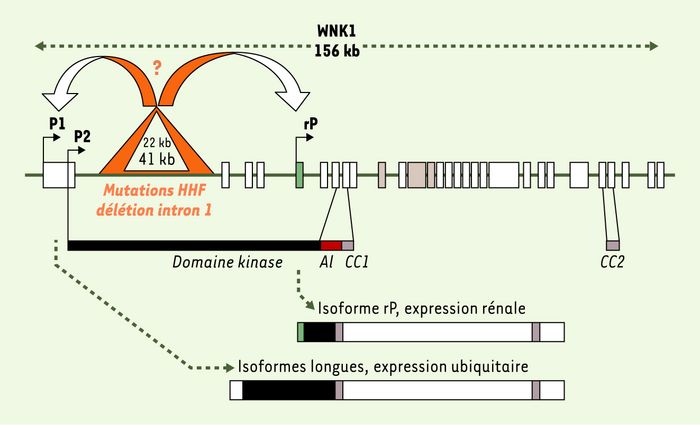
La structure exon/intron du gène humain est schématisée en haut de la figure. Les exons 9, 11 et 12 qui font l’objet d’épissage alternatif sont représentés par des rectangles gris ; l’exon 4a spécifique de l’isoforme rénale est indiqué par le rectangle vert. Trois promoteurs alternatifs P1, P2 et rP produisent deux types de transcrits : des isoformes longues contenant l’ensemble du domaine kinase et une isoforme rénale tronquée sans activité kinase. Les deux triangles (orange et blanc) représentent les délétions retrouvées dans l’intron 1 du gène chez deux familles atteintes. Ces délétions pourraient retentir sur l’expression de WNK1.
Le transcrit contenant l’ensemble des séquences codantes est majoritaire dans le coeur et le muscle squelettique. Cette isoforme longue est également exprimée très faiblement et de façon diffuse dans l’ensemble du rein. Une isoforme plus courte est fortement exprimée dans le rein, et plus spécifiquement dans le DCT et le CNT [9, 10]. Notre laboratoire a récemment caractérisé cette isoforme [9] : sa transcription débute au niveau de l’exon 4a sous le contrôle d’un promoteur spécifique (rP), donnant ainsi naissance à une protéine dépourvue des sous-domaines I à X du domaine kinase. Cette isoforme, spécifique du rein, ne possèderait donc pas d’activité kinase [5, 9]. Elle contient cependant toujours le domaine d’auto-inhibition situé en aval du domaine kinase. Ce domaine peut inhiber l’activité kinase de l’enzyme elle-même ou celle d’une kinase associée. On peut donc supposer que l’isoforme rénale de WNK1 pourrait inhiber l’activité kinase de WNK4 et, par conséquent, son action sur le NCC, mais cela reste à déterminer.
WNK4 et sécrétion de potassium
L’hyperkaliémie est souvent présente dans les acidoses tubulaires distales rénales. Son mécanisme n’était pas clair dans l’HHF, présenté comme la conséquence secondaire d’altérations de transfert des ions Na+ et Cl-, voire d’une altération de l’excrétion urinaire de K+. Toujours dans l’oeuf de xénope, K.T. Kahle et al. [19] ont montré que WNK4 est capable d’inhiber l’activité de ROMK, réduisant ainsi la sécrétion de potassium. Comme dans le cas de NCC, cette inhibition passe par la diminution du nombre de transporteurs présents dans la membrane (Figure 2). Plus précisément, WNK4 induirait l’endocytose clathrine-dépendante de ROMK et donc son retrait de la membrane. Cet effet ne semble pas dépendant de l’activité kinase de l’enzyme. En effet, une protéine WNK4 porteuse d’un domaine kinase muté est toujours capable d’inhiber l’activité de ROMK [19]. Ce dernier s’associerait, de façon directe ou indirecte, à un domaine situé en carboxyterminal du domaine kinase de WNK4.
Les mutations de WNK4 peuvent ainsi jouer par deux mécanismes : perte de l’inhibition sur NCC, augmentation de la capacité d’endocytose de ROMK et donc augmentation de l’activité de ces deux transporteurs (Figure 2). La rétention de sodium et de potassium serait donc exacerbée en présence des mutations HHF, ce qui est en accord avec la rétention hydrosodée et l’hyperkaliémie observées dans la maladie.
WNK4 et transport de chlore
Plusieurs hypothèses physiopathologiques ont été émises pour expliquer l’HHF. Il y a plus de 20 ans, M. Schambelan [20] décrivit celle d’un « shunt du chlore » : la perméabilité paracellulaire au chlore se trouverait augmentée chez les sujets atteints, entraînant une augmentation de la réabsorption de sodium sans modification de l’excrétion d’ions K+ et H+, d’où l’hyperkaliémie, l’hyperchlorémie avec acidose métabolique et hypervolémie.
Cette hypothèse a récemment été testée in vitro dans des cellules épithéliales de rein de chien (lignée MDCK) surexprimant la forme sauvage ou mutée (Asp564Ala) de WNK4. K. Yamauchi et al. [21] ont ainsi montré que seule la surexpression de la forme mutée augmente la perméabilité de ces cellules au chlore, à la suite d’une phosphorylation accrue des claudines 1-4 (Figure 2). Les claudines sont des protéines transmembranaires retrouvées au niveau des jonctions serrées dans de nombreux épithéliums et impliquées dans la régulation de la perméabilité paracellulaire. La colocalisation de WNK4 avec d’autres protéines d’expression membranaire, telles que ZO-1, au niveau des jonctions serrées du tubule rénal distal [3], est cohérente avec cette interaction. La famille des claudines contient au moins 20 membres. Chacun possède un profil d’expression spécifique. Les claudines 1-4, 8, 10, 11 et 16 sont exprimées dans le tubule rénal. K. Yamauchi et al. [21] ont montré que la surexpression de WNK4 sauvage ou muté ne modifie pas le niveau ni le profil d’expression intracellulaire des claudines 1-4 mais augmente leur phosphorylation. WNK4 pourrait ainsi agir comme un régulateur spécifique de la perméabilité paracellulaire au chlore, en augmentant sélectivement cette perméabilité, sans action sur la morphologie des jonctions serrées ou sur le transfert d’autres solutés non chargés [22].
Le rôle de WNK4 dans la régulation du transport de chlore semble aussi pouvoir s’exercer au niveau cellulaire. K.T. Kahle et al. [12] ont en effet montré, dans le système de l’oeuf de xénope, que WNK4 inhibe l’activité du transporteur basolatéral Na+-K+-Cl-, exprimé dans l’épithélium du pancréas, du côlon, des glandes sudoripares, de l’épididyme et du système respiratoire. Cette inhibition semble là aussi liée à la diminution du nombre de transporteurs présents dans la membrane. Les mêmes auteurs ont montré que WNK4 inhibe l’activité de CFEX, transporteur apical exprimé dans l’épithélium intestinal, pancréatique, testiculaire et hépatique, mais pas celle de la pendrine, autre transporteur apical exprimé au niveau de l’oreille interne, de la glande thyroïde et d’une sous-population de cellules intercalaires du canal collecteur.
Conclusions et perspectives
Ces études réalisées in vitro dans des oeufs de xénope ou des cellules rénales en culture ont permis de faire un premier lien entre WNK4 et transport du sodium, du potassium et du chlore, aussi bien dans le rein que dans des sites extra-rénaux. Il reste maintenant à démontrer que tel est le cas ex vivo, sur tubules isolés, et, in vivo, dans l’animal entier.
Une deuxième question importante est de caractériser le rôle de WNK1 dans la régulation de ces transporteurs. En effet, WNK1 et WNK4 pourraient interagir dans le DCT afin de régler positivement ou négativement l’activité de NCC [18]. Comme cela est discuté dans cet article, il reste à clarifier le rôle respectif de chacune des isoformes de WNK1 dans cette régulation. WNK1 étant exprimé dans les territoires où ROMK est exprimé, il est possible que WNK1 participe également à la régulation de la sécrétion de potassium. De plus, WNK1 et WNK4 étant co-exprimés dans les épithéliums impliqués dans la réabsorption du chlore, ils pourraient là aussi interagir afin de contrôler l’équilibre ionique.
L’originalité de cette nouvelle famille de kinases est de se situer au carrefour des régulations de transports ioniques. Au niveau du tubule rénal distal, WNK1 et WNK4 sont exprimés dans ce qui est défini comme l’ASDN (aldosterone sensitive distal nephron), à savoir le DCT, le CNT et le CCD. Leurs mécanismes de régulation, en particulier l’influence de l’aldostérone et/ou des protéines induites par l’aldostérone, sont une des nombreuses inconnues qui restent à élucider pour clarifier leur importance physiologique et l’intérêt éventuel d’un développement pharmacologique spécifique.
Appendices
Note
-
[1]
Maladie de transmission autosomique récessive associant une perte de sel avec alcalose hypokaliémique, hypomagnésémie et hypocalciurie, en rapport avec des mutations du gène SLC12A3 qui code pour le cotransporteur Na+-Cl- sensible aux diurétiques thiazidiques.
Références
- 1. Gordon RD, Klemm SA, Tunny TJ, et al. Gordon’s syndrome : a sodium-volume-dependent form of hypertension with a genetic basis. In : Laragh JH, Brenner BM, eds. Hypertension : pathophysiology, diagnosis, and management, 2nd ed. New York : Raven, Press Ltd, 1995 : 2111-3.
- 2. Disse-Nicodeme S, Desitter I, Fiquet-Kempf B, et al. Genetic heterogeneity of familial hyperkalaemic hypertension. J Hypertens 2001 ; 19 : 1957-64.
- 3. Wilson FH, Disse-Nicodeme S, Choate KA, et al. Human hypertension caused by mutations in WNK kinases. Science 2001 ; 293 : 1107-12.
- 4. Xu B, English JM, Wilsbacher JL, et al. WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J Biol Chem 2000 ; 275 : 16795-801.
- 5. Xu BE, Min X, Stippec S, Lee BH, et al. Regulation of WNK1 by an autoinhibitory domain and autophosphorylation. J Biol Chem 2002 ; 277 : 48456-62.
- 6. Verissimo F, Jordan P. WNK kinases, a novel protein kinase subfamily in multi-cellular organisms. Oncogene 2001 ; 20 : 5562-9.
- 7. Wang Z, Yang CL, Ellison DH. Comparison of WNK4 and WNK1 kinase and inhibiting activities. Biochem Biophys Res Commun 2004 ; 317 : 939-44.
- 8. Xu BE, Stippec S, Lenertz L, et al. WNK1 activates ERK5 by an MEKK2/3-dependent mechanism. J Biol Chem 2004 ; 279 : 7826-31.
- 9. Delaloy C, Lu J, Houot AM, et al. Multiple promoters in the WNK1 gene : one controls expression of a kidney-specific kinase-defective isoform. Mol Cell Biol 2003 ; 23 : 9208-21.
- 10. O’Reilly M, Marshall E, Speirs HJ, Brown RW. WNK1, a gene within a novel blood pressure control pathway, tissue-specifically generates radically different isoforms with and without a kinase domain. J Am Soc Nephrol 2003 ; 14 : 2447-56.
- 11. Choate KA, Kahle KT, Wilson FH, et al. WNK1, a kinase mutated in inherited hypertension with hyperkalemia, localizes to diverse Cl--transporting epithelia. Proc Natl Acad Sci USA 2003 ; 100 : 663-8.
- 12. Kahle KT, Gimenez I, Hassan H, et al. WNK4 regulates apical and basolateral Cl- flux in extrarenal epithelia. Proc Natl Acad Sci USA 2004 ; 101 : 2064-9.
- 13. Schnermann J. Sodium transport deficiency and sodium balance in gene-targeted mice. Acta Physiol Scand 2001 ; 173 : 59-66.
- 14. Masilamani S, Wang X, Kim GH, et al. Time course of renal Na-K-ATPase, NHE3, NKCC2, NCC, and ENaC abundance changes with dietary NaCl restriction. Am J Physiol Renal Physiol 2002 ; 283 : 648-57.
- 15. Wang W. Regulation of renal K transport by dietary K intake. Annu Rev Physiol 2004 ; 66 : 547-69.
- 16. Yoo D, Kim BY, Campo C, et al. Cell surface expression of the ROMK (Kir 1.1) channel is regulated by the aldosterone-induced kinase, SGK-1, and protein kinase A. J Biol Chem 2003 ; 278 : 23066-75.
- 17. Wilson FH, Kahle KT, Sabath E, et al. Molecular pathogenesis of inherited hypertension with hyperkalemia : the Na-Cl cotransporter is inhibited by wild-type but not mutant WNK4. Proc Natl Acad Sci USA 2003 ; 100 : 680-4.
- 18. Yang CL, Angell J, Mitchell R, Ellison DH. WNK kinases regulate thiazide-sensitive Na-Cl cotransport. J Clin Invest 2003 ; 111 : 1039-45.
- 19. Kahle KT, Wilson FH, Leng Q, et al. WNK4 regulates the balance between renal NaCl reabsorption and K+ secretion. Nat Genet 2003 ; 35 : 372-6.
- 20. Schambelan M, Sebastian A, Rector FC Jr. Mineralocorticoid-resistant renal hyperkalemia without salt wasting (type II pseudohypoaldosteronism) : role of increased renal chloride reabsorption. Kidney Int 1981 : 19 : 716-27.
- 21. Yamauchi K, Rai T, Kobayashi K, et al. Disease-causing mutant WNK4 increases paracellular chloride permeability and phosphorylates claudins. Proc Natl Acad Sci USA 2004 ; 101 : 4690-4.
- 22. Kahle KT, MacGregor GG, Wilson FH, et al. Paracellular Cl– permeability is regulated by WNK4 kinase : Insight into normal physiology and hypertension. Proc Natl Acad Sci USA 2004 ; 101 : 14877-82.
List of figures
Figure 1
Localisations chromosomiques, structures géniques et domaines fonctionnels conservés des WNK (with no lysine K kinase).
Les gènes correspondant aux locus PHA2B (17p11-q21) et PHA2C (12p13.3) sont des homologues : WNK1 et WNK4 appartiennent à la famille des sérine-thréonine kinases WNK, caractérisée par l’absence de la lysine active dans le sous-domaine II du domaine kinase. La comparaison des séquences nucléiques (partie gauche de la figure) et protéiques (partie droite) met en évidence trois zones d’homologie. Le domaine catalytique sérine-thréonine kinase, le domaine auto-inhibiteur et les deux domaines coiled-coil (hélices hydrophobes) sont conservés.
Figure 2
Transport du Cl-, du Na+ et du K+ dans le néphron distal et hypothèses physiopathologiques suggérées dans l’hypertension hyperkaliémique familiale (HHF).
Représentation schématique du néphron et des cellules qui composent le tubule contourné distal (DCT) et le canal collecteur cortical (CCD). Ce dernier est constitué de trois types cellulaires : les cellules principales et les cellules intercalaires de types A et B figurées de haut en bas. Les pompes sont représentées en bleu, les échangeurs en orange, les cotransporteurs en violet et les canaux en vert. Le récepteur minéralocorticoïde (MR) est représenté en jaune, les claudines 1-4 en bleu dans le complexe de jonction serrée représenté en rose. À gauche, sont schématisés les mécanismes d’action des WNK suggérés par les expériences réalisées dans les oeufs de xénope et les cellules rénales en culture. À droite, sont schématisées les conséquences des mutations WNK4. Ces différents mécanismes physiopathologiques aboutissent à une augmentation de la réabsorption de Na+ et Cl- et une diminution de la sécrétion de K+ expliquant les anomalies biologiques (hyperkaliémie, hyperchlorémie) et l’hypertension artérielle . CNT : tubule connecteur ; CCM : canal collecteur médullaire ; NCC : cotransporteur Na+Cl- apical sensible aux diurétiques thiazidiques ; ROMK : canal potassique apical.
Figure 3
Mutations de WNK4 à l’origine de l’hypertension hyperkaliémique familiale (HHF).
La structure exon/intron du gène humain est schématisée en haut de la figure, ainsi que le domaine kinase, le domaine auto-inhibiteur (AI) et les domaines coiled coil (CC1 et CC2). Les quatre mutations faux-sens identifiées dans plusieurs familles atteintes d’HHF sont localisées au niveau de courts motifs conservés de 10 et 16 acides aminés (aa) en aval des domaines coiled coil. Chacune de ces mutations modifie un résidu conservé entre les quatre membres de la famille WNK.
Figure 4
Mutations de WNK1 à l’origine de l’hypertension hyperkaliémique familiale (HHF).
La structure exon/intron du gène humain est schématisée en haut de la figure. Les exons 9, 11 et 12 qui font l’objet d’épissage alternatif sont représentés par des rectangles gris ; l’exon 4a spécifique de l’isoforme rénale est indiqué par le rectangle vert. Trois promoteurs alternatifs P1, P2 et rP produisent deux types de transcrits : des isoformes longues contenant l’ensemble du domaine kinase et une isoforme rénale tronquée sans activité kinase. Les deux triangles (orange et blanc) représentent les délétions retrouvées dans l’intron 1 du gène chez deux familles atteintes. Ces délétions pourraient retentir sur l’expression de WNK1.