Article body
Chaque année en France, la iatrogénie médicamenteuse est responsable d’environ 128 000 hospitalisations, pour un coût global estimé à 320 millions d’euros et l’incidence des hospitalisations liées à un effet indésirable d’un médicament est de 3,2 % [1]. Ces chiffres démontrent que les variations individuelles de réponse aux médicaments représentent un problème médical et de Santé Publique important. La pharmacogénétique étudie les mécanismes d’origine génétique intervenant dans la réponse aux médicaments avec pour objectif l’optimisation des traitements, aussi bien en termes d’efficacité que de sécurité d’emploi. Dans cet article, nous allons exposer les « bases » de l’émergence de la pharmacogénétique comme outil de prescription thérapeutique en citant quelques exemples d’applications cliniques courantes.
Polymorphismes génétiques et susceptibilité aux médicaments
Le séquençage complet du génome humain et la mise au point de technologies performantes d’analyse des gènes a permis l’identification de variations de séquences dans les gènes cibles des médicaments. La pharmacogénétique s’intéresse aux conséquences de ces polymorphismes génétiques en thérapeutique dans le but de développer des tests simples permettant d’identifier les individus susceptibles de présenter des anomalies de réponse (inefficacité, toxicité). La plupart des gènes (codant pour des protéines intervenant dans le métabolisme, le transport, les récepteurs, la transduction du signal) sont susceptibles d’avoir un rôle déterminant dans la réponse aux médicaments. Cependant les gènes les plus étudiés sont ceux dont les produits interviennent dans le métabolisme et le transport de molécules thérapeutiques [2, 3].
Les polymorphismes génétiques peuvent être responsables de variations d’expression ou d’activité des enzymes du métabolisme des médicaments. Ils s’expriment dans la population générale sous la forme de différents phénotypes métaboliques, définissant généralement deux groupes d’individus : métaboliseurs lents (ML) (déficit d’activité enzymatique) et métaboliseurs rapides (MR) (activité normale). L’existence de métaboliseurs ultrarapides (MUR) (activité augmentée) ou intermédiaires (MI) (activité réduite) est possible pour certaines enzymes. La fréquence des différents phénotypes est variable en fonction de l’enzyme, et, pour une même enzyme, variable en fonction de l’origine ethnique ou géographique des populations étudiées. Les conséquences cliniques des polymorphismes génétiques dépendent de plusieurs facteurs : importance de la voie métabolique polymorphe dans la clairance globale du médicament, administration du médicament sous forme active ou de pro-drogue, activité des métabolites.
Prédiction de la réponse aux médicaments : phénotypage ou génotypage
Les méthodes de phénotypage reposent soit sur une mesure de l’activité enzymatique soit sur l’administration d’un substrat-test (en général un médicament), suivie d’une mesure des quantités résiduelles de substrat et/ou de leurs métabolites à partir d’un échantillon biologique, urinaire ou sanguin. On détermine le rapport métabolique entre la quantité de substance retrouvée sous forme inchangée et celle d’un (ou plusieurs) métabolite (s), rapport qui est le reflet de l’activité enzymatique étudiée. Les méthodes de phénotypage présentent certains inconvénients (absence d’un substrat-test présentant toutes les qualités requises, spécificité, innocuité ou contre-indications) qui limitent leur utilisation. En pratique, le génotypage est plus largement utilisé que le phénotypage puisqu’il est applicable à l’analyse de l’ensemble des polymorphismes affectant non seulement la pharmacocinétique des médicaments, mais également leurs effets (récepteurs, cibles protéiques). Des études de corrélation phénotype/génotype, complétées parfois par la caractérisation fonctionnelle in vitro des variants, sont un préalable nécessaire à l’utilisation de ces tests.
Applications cliniques
Parmi les exemples importants, on peut citer le CYP 2D6 (CY : cytochrome), impliqué dans le métabolisme de plus d’une centaine de médicaments (anti-arythmiques, β-bloquants, psychotropes, dérivés opiacés à visée analgésique ou antitussive), soit 20 à 25 % de l’ensemble des médicaments d’usage courant et d’intérêt thérapeutique majeur (Tableau I) [4]. Des adaptations de la posologie des antidépresseurs en fonction du phénotype sont proposées par de nombreux auteurs qui recommandent des doses pouvant aller de 30 % de la dose standard conventionnelle pour les ML, à plus de 200 % pour les MUR [5]. Le cas de la thiopurine méthyl-transférase (TPMT), enzyme responsable de l’élimination de la 6-mercaptopurine et de l’azathioprine, illustre particulièrement bien l’intérêt de la pharmacogénétique [6]. Des variants génétiques de la TPMT ont été associés à la survenue de toxicités hématologiques sévères chez des patients traités. Ainsi, des doses réduites doivent être prescrites aux sujets ayant un déficit en cette enzyme et à l’inverse, les sujets ayant une activité élevée doivent recevoir des doses plus fortes pour obtenir une efficacité thérapeutique. En cancérologie, le dépistage de la maladie de Gilbert (déficit partiel en glucuroconjugaison) est effectué lorsque l’on envisage un traitement par l’irinotécan[1] pour éviter des toxicités hématologiques ou digestives très graves [7]. Le développement de la pharmacogénétique intéresse directement d’autres classes thérapeutiques, par exemple les anticoagulants, les immunosuppresseurs ou les anti-protéases [8-10].
Tableau I
Exemples de polymorphismes génétiques affectant la réponse aux médicaments.
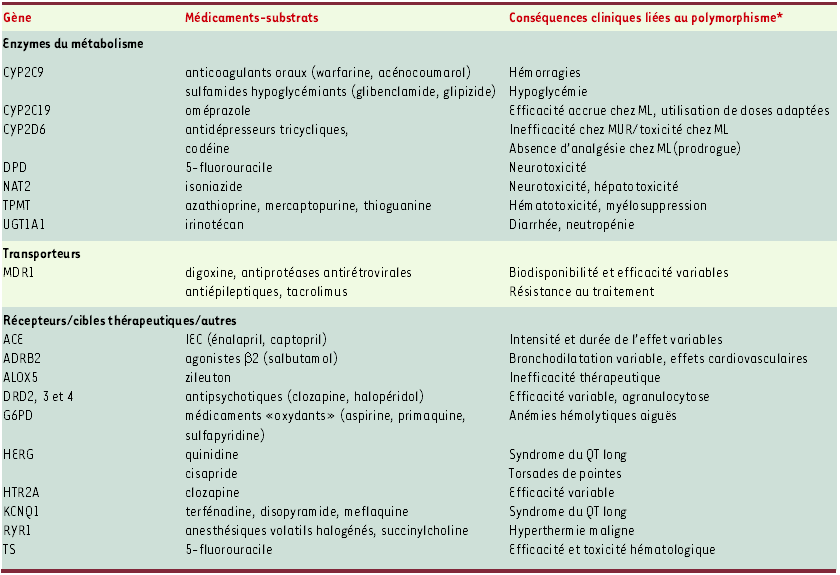
* Certaines corrélations restent controversées dans la littérature ou à confirmer sur un plus grand nombre de patients. CYP, cytochromes P450 ; DPD, dihydropyrimidine déshydrogénase ; NAT2, N-acétyltransférase 2 ; TPMT, thiopurine S-méthyltransférase ; UGT, uridine diphosphate glucuronosyltransférase ; MDR1, multi-drug resistance (ou ABCA1) ; ACE, enzyme de conversion de l’angiotensine ; ADRB2, récepteur β2-adrénergique ; DRD2, 3 et 4, récepteurs dopaminergiques D2, D3 et D4 ; HERG, KCNQ1, canaux potassiques ; HTR2A, récepteur sérotoninergique 5-HT2A ; G6PD, glucose 6-phosphate déshydrogénase ; RYR1, récepteur à la ryanodine ; TS, thymidylate synthase. ML : métaboliseurs lents ; MUR : métaboliseurs ultrarapides.
Perspectives
La prescription thérapeutique individualisée sur la base de facteurs génétiques semble aujourd’hui devenir une réalité compte tenu des progrès dans la connaissance des conséquences fonctionnelles des polymorphismes, l’identification des cibles médicamenteuses et le développement de technologies performantes de génotypage (rapides et peu coûteuses). Des études cliniques prospectives sur de larges cohortes doivent maintenant être menées non seulement pour démontrer l’importance des polymorphismes génétiques pour la prédiction de l’efficacité et de la toxicité des médicaments, mais également pour démontrer le bénéfice des tests pharmacogénétiques en terme d’économie de la santé.
Appendices
Note
-
[1]
Molécule cytotoxique utilisée en cancérologie agissant par l’inhibition spécifique de l’ADN topo-isomérase 1, enzyme essentielle à la réplication de l’ADN métabolisé partiellement par les carboxyestérases en un métabolite, le SN 38, inhibiteur plus actif que l’irinotécan, principal responsable de l’activité.
Références
- 1. Rapport de l’Agence française de sécurité sanitaire des produits de santé. Paris : AFSSAPS, 2001.
- 2. Evans WE, Relling MV. Pharmacogenomics : translating functional genomics into rational therapeutics. Science 1999 ; 286 : 487-91.
- 3. Oscarson M. Pharmacogenetics of drug metabolising enzymes : importance for personalised medecine. Clin Chem Lab Med 2003 ; 41 : 573-80.
- 4. Zanger UM, Raimundo S, Eichelbaum M. Cytochrome P450 2D6 : overview and update on pharmacology, genetics, biochemistry. Naunyn-Schmiedeberg’s Arch Pharmacol 2004 ; 369 : 23-37.
- 5. Kirchheiner J, Brosen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for anti-depressants : a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001 ; 104 : 173-92.
- 6. Relling MV, Hancock ML, River S, et al. Mercaptopurine therapy intolerance and heterozygoty at the thiopurine S-methyl-transferase gene locus. J Natl Cancer Inst 1999 ; 91 : 2001-8.
- 7. Ando Y, Saka H, Ando M, et al. Polymorphisms of UDP-glucuronosyl-transferase gene and irinotecan toxicity : a pharmacogenetics analysis. Cancer Res 2000, 60 : 6921-6.
- 8. Daly AK, King BP. Pharmacogenetics of oral anticoagulants. Pharmacogenetics 2003 ; 13 : 247-52.
- 9. Anglicheau D, Verstuyft C, Laurent-Puig P, et al. Association of the multidrug resistance-1 gene single-nucleotide polymorphisms with the tacrolimus dose requirements in renal transplant recipients. J Am Soc Nephrol 2003, 14 : 1889-96.
- 10. Swiss HIV Cohort Study. Response to antiretroviral treatment in HIV-1-infected individuals with allelic variants of the multidrug resistance transporter 1 : a pharmacogenetics study. Lancet 2002 ; 359 : 30-6.
List of tables
Tableau I
Exemples de polymorphismes génétiques affectant la réponse aux médicaments.
* Certaines corrélations restent controversées dans la littérature ou à confirmer sur un plus grand nombre de patients. CYP, cytochromes P450 ; DPD, dihydropyrimidine déshydrogénase ; NAT2, N-acétyltransférase 2 ; TPMT, thiopurine S-méthyltransférase ; UGT, uridine diphosphate glucuronosyltransférase ; MDR1, multi-drug resistance (ou ABCA1) ; ACE, enzyme de conversion de l’angiotensine ; ADRB2, récepteur β2-adrénergique ; DRD2, 3 et 4, récepteurs dopaminergiques D2, D3 et D4 ; HERG, KCNQ1, canaux potassiques ; HTR2A, récepteur sérotoninergique 5-HT2A ; G6PD, glucose 6-phosphate déshydrogénase ; RYR1, récepteur à la ryanodine ; TS, thymidylate synthase. ML : métaboliseurs lents ; MUR : métaboliseurs ultrarapides.