Abstracts
Résumé
Alors que la diffusion épidémique des virus de l’immunodéficience humaine (VIH) s’accompagne d’une mortalité très élevée, l’infection par les virus de l’immunodéficience simienne (SIV), retrouvée chez de nombreuses espèces de singes d’Afrique avec une forte prévalence, ne produit aucune maladie détectable. Les facteurs responsables de cette différence entre l’être humain et les porteurs naturels de SIV restent mal identifiés. Il est possible que cette différence ne réside ni dans les facteurs viraux, ni dans les facteurs d’hôte, mais dans la nature même de la relation hôte/virus, résultat d’un équilibre évolutif stable établi depuis longtemps dans certaines populations simiennes.
Summary
While the AIDS epidemic caused by human immunodeficiency viruses (HIV) has resulted in the death of over 20 million people worldwide, simian immunodeficiency virus (SIV) infection, found in numerous African primate species, does not induce disease symptoms. The factors accounting for this difference between humans and natural host of SIV remain poorly understood. The entangled nature of the host/virus relationship could be the answer, rather than independent virus or host factors. Such a relationship is as a consequence of host/virus adaptation which has evolved over long periods in naturally infected primate species.
Article body
Infection des primates par les lentivirus : une histoire ancienne
Plus d’une trentaine d’espèces différentes de primates africains sont aujourd’hui connues pour être naturellement infectées par une souche de SIV (simian immunodeficiency virus) [1, 2]. Contrairement à d’autres rétrovirus, les SIV ne sont retrouvés qu’au sein d’espèces d’Afrique. Ils sont classés en six lignées phylogénétiques distinctes [3] (Figure 1) ; cependant, la caractérisation d’un nombre de plus en plus important de virus différents a souligné que les relations phylogénétiques entre ces lentivirus de primates ne peuvent être interprétées de manière simple. En effet, certains de ces virus ne peuvent pas être classés parmi les six groupes précédemment définis, car ils possèdent des génomes hybrides ou mosaïques, signes de recombinaison entre souches de différentes lignées ; c’est le cas, entre autres, du SIVmnd-2 des mandrills et du SIVgsn des singes hocheurs [4, 5]. Pour expliquer ces données génétiques, deux modes principaux d’évolution, avec ou sans transfert interespèces, peuvent être proposés.
Figure 1
Arbre phylogénétique des lignées de SIV (simian immunodeficiency virus).
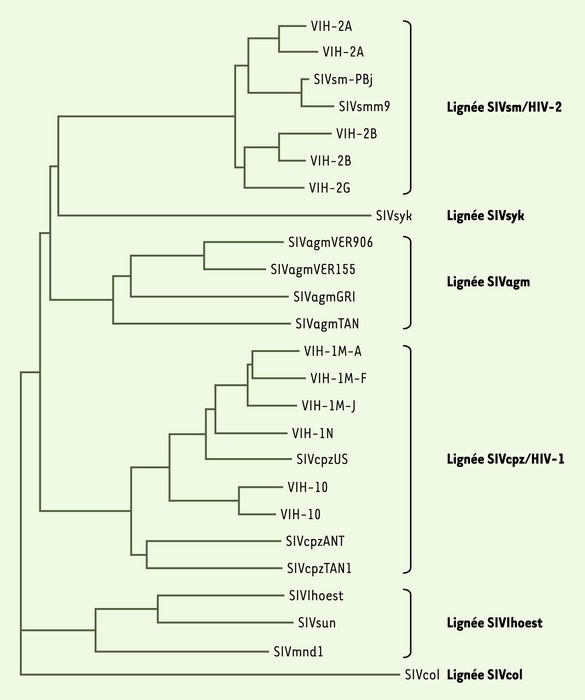
Cet arbre a été construit à partir des séquences protéiques Pol de lentivirus de primates représentatifs des six lignées principales : SIVcpz, retrouvée chez les chimpanzés (Pan troglodytes) et associée au VIH-1 ; SIVsm, identifiée chez le mangabey enfumé (Cercocebus atys) et associée au VIH-2 ; SIVagm, présente dans les quatre espèces de singes verts (membres du genre Chlorocebus) ; SIVlhoest, du singe de l’Hoesti (Cercopithecus lhoesti), associée au SIVsun du cercopithèque à queue de soleil (Cercopithecus solatus) et au SIVmnd-1 des mandrills (Mandrillus sphinx) ; SIVsyk, du cercopithèque à diadème (Cercopithecus albogularis) ; SIVcol, décrite chez le colobe guéréza (Colobus guereza). Cet arbre a été obtenu par la méthode neighbor joining, et sa robustesse évaluée par un test de bootstrap (100 répliques). La longueur des branches est proportionnelle à la distance évolutive entre les différentes souches virales.
La première situation est celle d’une coévolution hôte/virus : les virus, présents chez un ancêtre commun des singes infectés, ont divergé parallèlement à leurs espèces hôtes, sans transfert interespèces. On observe en faveur de cette hypothèse le fait que chaque espèce de primates, à l’exception notable du mandrill et de l’être humain, chez lesquels deux types de virus sont retrouvés, ne semble infectée que par une seule lignée de virus. De plus, la phylogénie des virus est parallèle à celle des espèces hôtes. Ainsi, la superespèce singe vert est divisée en quatre espèces principales selon des critères morphologiques, génétiques et de répartition géographique, et chacune est porteuse d’un SIVagm distinct, désigné SIVagmVer, SIVagmGri, SIVagmTan ou SIVagmSab selon l’espèce à partir de laquelle il a été isolé.
À l’opposé, lorsque la parenté virale suit la proximité géographique plutôt que phylogénétique des espèces hôtes, un transfert interespèces est proposé. Ainsi, plusieurs transmissions interespèces, plus ou moins anciennes, semblent requises pour expliquer la position des souches humaines VIH-1 et -2 au sein de groupes bien distincts de virus (Figure 1) : ces deux types de virus humains ressemblent aux virus simiens présents dans la même zone géographique. De même, le SIVmnd-2, présent seulement au nord du fleuve Ogoue (Gabon), diffère du SIVmnd-1 retrouvé chez les mandrills vivant au sud du fleuve, mais il appartient à la même lignée que le SIVdrl, présent chez les drills, et dont l’habitat voisine avec celui des mandrills porteurs du SIVmnd-2 [5].
Pouvoir pathogène des lentivirus
Malgré un taux élevé d’infection (séroprévalence allant de 20 % à 60 %) dans les populations adultes sauvages comme celles des mangabeys enfumés, des singes verts ou des mandrills [6], ces infections par lentivirus sont naturellement asymptomatiques chez leur hôte naturel. Cependant, dans certains cas, l’infection est pathogène.
Variations du pouvoir pathogène
Différents exemples d’infections pathogènes par des lentivirus peuvent être cités, dont le plus évident est bien sûr celui de VIH-1 et -2, agents étiologiques du sida humain. Il est généralement admis que les virus VIH-1 et -2 résultent respectivement d’une transmission zoonotique du SIVcpz de chimpanzés (Pan troglodytes) et du SIVsm de mangabeys enfumés (sooty mangabey, Cercocebus atys) [1]. Le caractère accidentel et relativement récent de l’infection humaine est souvent mis en avant pour expliquer la pathogénicité des VIH. L’épidémie d’infection par le VIH-1 groupe M (M pour main) s’est largement répandue à travers le monde, tandis que l’infection par le VIH-2 est restée centrée sur l’Afrique de l’ouest. Le caractère plus endémique et peut-être ancien de l’infection par le VIH-2 dans les populations d’Afrique occidentale pourrait expliquer une transmissibilité, une virulence et une progression de la maladie plus faibles que celles de la maladie déclenchée par le VIH-1, même si le tableau clinique au stade sida, lorsqu’il survient, est similaire [7].
Un autre exemple d’acquisition de virulence au cours d’une transmission interespèces récente est celui du SIV du macaque. De nombreuses souches de SIVmac ont été isolées chez différentes espèces de macaques (à queue de cochon, Rhésus ou Cynomolgus) en captivité, présentant des signes cliniques semblables à ceux du sida ; pourtant, dans leur habitat naturel, l’Asie, ils ne semblent pas infectés par ces virus, le SIVmac n’étant jamais retrouvé chez le macaque en milieu naturel. Il est fort probable que le SIVmac provienne d’une transmission du SIVsm de mangabeys enfumés à des macaques vivant dans les mêmes centres de primatologie, cette transmission interespèces déterminant une infection pathogène chez ces hôtes « accidentels ». De fait, l’infection expérimentale de macaques par des souches de SIVmac/sm se traduit presque toujours par l’apparition d’un sida (pour revue, voir [8]).
Un cas exceptionnel est représenté par l’isolat SIVsmm-PBj14, produit par l’inoculation de l’isolat SIVsm chez un macaque et le réisolement du virus, 14 mois plus tard, chez l’animal alors en phase de sida. Le virus réisolé présente de nouvelles propriétés : il provoque une maladie très rapidement fatale (1 à 2 semaines) chez le macaque, mais également chez le mangabey enfumé séronégatif, qui est pourtant l’hôte naturel du SIVsm [9]. Dans ce cas, il semble que le passage chez un hôte « accidentel » comme le macaque ait sélectionné des variations virales conférant une virulence vis-à-vis de l’espèce hôte d’origine.
Une situation quelque peu différente est celle de l’infection par le SIVagm, asymptomatique chez son hôte naturel, le singe vert, mais également après transfert interespèce chez les macaques Rhésus ou Cynomolgus. En revanche, l’infection expérimentale par un isolat particulier, le SIVagm-ver90, entraîne le développement d’une immunodéficience chez une seule espèce, le macaque à queue de cochon [10]. On voit ainsi que le changement d’espèce ne produit pas systématiquement un caractère pathogène.
Un dernier exemple dans ce sens est celui des chimpanzés infectés expérimentalement par le VIH-1. L’établissement d’une infection chronique durable a été mise en évidence, mais seul un chimpanzé infecté par deux souches de VIH-1 a développé un sida au bout de 9 ans [11].
Ces exemples suggèrent qu’il existe deux profils d’infection lentivirale chez les primates : une infection asymptomatique, se maintenant de manière endémique dans la population, et une évolution pathologique chez certaines espèces vraisemblablement exposées plus récemment à l’infection lentivirale.
Fondements génétiques de la virulence
Conformément aux notions classiques, un phénotype très réplicatif ex vivo en culture de cellules est souvent le fait de souches peu pathogènes in vivo. Même si on ne sait pas encore pourquoi, il est clair qu’il n’y a pas de corrélation entre la réplication in vitro et la virulence in vivo : les fondements génétiques de la virulence doivent donc être étudiés in vivo. On utilise pour cela des virus porteurs de génomes mutants ou chimériques, ces derniers comportant une partie issue d’un virus pathogène dans le contexte d’un virus non pathogène, ou l’inverse. De telles études doivent être menées sur des groupes d’animaux de taille suffisante, afin de contrôler les variations individuelles. L’ensemble de ces expérimentations est très lourd au regard des considérations éthiques, du coût des modèles simiens et des durées nécessaires à l’observation de signes de sida.
Les mutations virales introduites expérimentalement pour rechercher les facteurs de virulence sont de deux catégories : celles entraînant une perte de fonction virulente et celles entraînant un gain de virulence. Les études de perte de virulence ont souvent été motivées par la recherche de vaccins vivants atténués. Ainsi, la faible charge virale et l’absence d’effets pathogènes après l’infection de macaques par un virus SIVmac ayant subi une délétion expérimentale du gène nef, alors que le SIVmac porteur de nef est toujours pathogène, a suggéré que la protéine nef constituait un facteur de virulence [12]. Ces observations ont été renforcées par l’isolement, chez un porteur humain asymptomatique à long terme, d’un virus VIH-1 porteur d’une délétion spontanée dans le gène nef [13]. Cependant, tous les SIV d’Afrique identifiés possèdent une phase de lecture ouverte correspondant à une protéine Nef fonctionnelle. La fonction nef, si elle est certainement indispensable à une réplication efficace in vivo, ne peut donc à elle seule expliquer les différences de virulence virale observées chez les primates non humains.
Dans le sens d’un gain de fonction, certaines mutations ont montré une capacité à accroître la virulence du SIVmac : éléments enhancers additionnels, sites de glycosylation supplémentaires dans l’enveloppe (boucle V1) ou capacités d’activation cellulaire de nef permettant la réplication virale dans des cellules non stimulées (voir par exemple [14-18]). Cependant, ces mutations ne produisent de virulence que dans les contextes génétiques où toutes les autres mutations sont déjà présentes : aucune ne constitue donc à elle seule un déterminant de virulence.
De nombreux travaux ont également porté sur les fondements moléculaires de la virulence d’hybrides SIV/VIH, appelés SHIV (simian human immunodeficiency virus), permettant la conduite d’essais de vaccins dérivés d’antigènes du VIH chez le macaque. Dans ce modèle, divers déterminants génétiques, notamment de l’enveloppe, ont pu être associés au pouvoir pathogène (voir [19] pour des références récentes).
Comme pour d’autres agents pathogènes, viraux ou autres, il n’a donc pas été possible d’identifier un facteur de virulence unique. On peut presque conclure que la virulence du SIV nécessite la présence de nombreux éléments génétiques dans un environnement d’hôte particulier, ce qui revient malheureusement à ne pas expliquer grand-chose.
Tropisme viral et pouvoir pathogène
L’identification des récepteurs d’entrée des virus (famille des récepteurs des chimiokines) et la mise en évidence de leur diversité selon les souches virales a encouragé la recherche d’une association entre certains tropismes cellulaires et pouvoir pathogène. Cette hypothèse a été soutenue par l’observation des variations du tropisme du VIH au cours de l’histoire naturelle de l’infection humaine : durant la phase de séropositivité asymptomatique, les virus détectés sont du groupe tropique R5, à préférence « macrophagique », alors que la phase clinique symptomatique voit souvent le développement de souches du groupe tropique X4, à préférence « lymphocytaire ». Dans le même sens, la plupart des souches de SIV naturels, ainsi que le VIH-2, appartiennent plutôt au groupe R5 (voir par exemple [20]).
Il pouvait alors être proposé que la capacité d’utilisation virale du récepteur X4 (CXCR-4) soit indispensable à l’induction de l’immunodéficience [19]. Cependant, seulement 50 % des individus progressant vers le sida présentent de façon prédominante des virus X4, et les virus SIVmac sont, tout au long de l’infection chez le macaque, de type R5 et non pas X4, et induisent pourtant un sida. Même si le débat n’est, pour la plupart des auteurs, ni simple ni clos, l’acquisition du tropisme X4 semble être une conséquence de l’immunodéficience plutôt qu’une cause. En l’absence de réponse immune, comme c’est le cas en culture cellulaire, une capacité réplicative élevée serait sélectionnée, qui favoriserait le tropisme X4, permettant en cela l’accès du virus aux lymphocytes activés capables de soutenir une réplication virale abondante [21].
Charge virale et pouvoir pathogène
En pratique médicale, la mesure de charge virale représente, avec le comptage des lymphocytes T CD4 périphériques, un marqueur de choix du suivi de l’évolution de la maladie. Ainsi, les sujets asymptomatiques présentent généralement une charge virale réduite, qui constitue d’ailleurs l’un des critères définissant la situation clinique de « non-progression à long terme ». Il était raisonnable de proposer que la capacité de l’organisme à contrôler la charge virale, notamment par une réponse immune efficace, soit la clé des différences de virulence. L’infection serait alors conçue comme un bras de fer entre le virus et l’organisme, et la victoire de l’organisme sur le virus expliquerait la non-virulence de certaines infections.
Cette hypothèse semble satisfaisante en première analyse : dans les situations où il y a peu de virus, il est logique d’attendre moins de conséquences pathologiques. Cependant, une telle explication ne tient pas dans le cadre des infections non pathogènes naturelles. Théoriquement, l’adaptation hôte-virus doit permettre une survie (et une reproduction) maximale de l’hôte, mais aussi une réplication maximale du virus. De plus, la capacité adaptative des lentivirus des primates est exceptionnelle et plus rapide que celle de leur hôte : il suffit pour s’en convaincre d’observer les capacités de résistance aux antiviraux, d’échappement immunologique ou d’adaptation des lentivirus à des types cellulaires variés. Peut-on imaginer que l’organisme des singes adaptés au SIV ait trouvé une solution que le virus ne peut définitivement pas contourner ? L’adaptation réciproque réussie n’est-elle pas plutôt définie par une situation où le virus se réplique le plus efficacement possible sans affecter la reproduction de son hôte ? Conformément à cette idée, les études ont révélé des charges virales élevées chez les singes porteurs naturels asymptomatiques [6]. De plus, l’équilibre évolutif hôte-virus paraît stable. La population virale ne subit pas de fortes contraintes chez les hôtes simiens naturels en situation non pathogène : une grande diversité et une grande taille des populations virales, reflets des charges virales élevées, s’accompagnent de faibles pressions immunologiques en faveur du changement [22, 23].
Mécanismes de l’adaptation hôte-virus
L’existence de charges virales plasmatiques élevées chez les singes asymptomatiques a poussé à examiner de manière plus détaillée les conséquences de l’infection, notamment sur la prolifération et le renouvellement des lymphocytes T. Alors qu’une prolifération anormale des lymphocytes T, associée à des phénomènes d’apoptose, a pu être mesurée dans les espèces, comme le macaque Rhésus [24], chez lesquelles le SIV est pathogène, prolifération et apoptose sont absentes, à souche et charge virale identique, chez les hôtes naturels [25]. L’absence de prolifération prolongée des cellules T CD8+ pourrait traduire une baisse d’immunogénicité du virus chez son hôte naturel, par des mécanismes qui sont toutefois encore mal compris. Cette situation constituerait une forme d’échappement immunologique stable, ne nécessitant pas de variation antigénique continuelle. La réduction de l’activation immunitaire bénéficierait certes au virus, mais aussi à l’hôte, en diminuant les phénomènes immunopathologiques liés aux destructions tissulaires et au renouvellement rapide des cellules T [26, 27]. Finalement, l’adaptation hôte-virus se définirait par une modification de l’équilibre entre la réplication du virus et sa destruction immunologique, modification résultant d’une diminution adaptative de l’immunogénicité du virus chez son hôte naturel. Dans ce sens, si l’on considère que le SIV est très immunogène chez le macaque, la charge virale mesurée résulte d’une réplication virale abondante, mais aussi d’une destruction immunologique intense du virus. Chez l’hôte naturel, la charge virale observée reste élevée, conséquence d’une réplication virale équivalente, mais d’une destruction immunologique moins efficace. En raison de cette réaction immunologique plus faible, l’hôte naturel ne subit les conséquences ni de l’activation chronique, ni de la destruction des tissus infectés.
La stabilité d’une telle relation étant très dépendante des caractéristiques immunologiques individuelles de l’hôte, cet équilibre serait facilement rompu lors de passage interespèces. Il faut noter qu’un tel mécanisme d’accroissement de la virulence ne passe pas nécessairement par un changement d’espèce. Chez l’être humain, un déséquilibre de la relation hôte-virus aurait pu survenir lors de la diffusion épidémique du virus dans des populations bien différentes sur le plan immunologique, expliquant ainsi que le VIH-1, épidémique, semble globalement plus pathogène que le VIH-2, resté plus endémique.
Conclusions
Les perturbations introduites par l’homme dans son milieu naturel (déforestation, mais surtout urbanisation…) ont fourni les conditions épidémiologiques provoquant la pandémie de sida. La pandémie, à son tour, a modifié les équilibres entre les virus et leurs hôtes naturels, révélant des propriétés pathogènes nouvelles. L’étude des infections SIV non pathogènes chez leurs hôtes naturels éclaire le caractère ambigû, à la fois bénéfique et dangereux, de la réponse immune antivirale, ainsi que les mécanismes de la virulence du VIH.
Appendices
Remerciements
À Eric Delaporte et Martine Peteers (IRD, Montpellier), ainsi qu’à l’ANRS et à Sidaction-Ensemble contre le sida.
Références
- 1. Hahn BH, Shaw GM, De Cock KM, Sharp PM. AIDS as a zoonosis : scientific and public health implications. Science 2000 ; 287 : 607-14.
- 2. Peeters M, Courgnaud V, Abela B, et al. Risk to human health from a plethora of simian immunodeficiency viruses in primate bushmeat. Emerg Infect Dis 2002 ; 8 : 451-7.
- 3. Peeters M, Courgnaud V, Abela B. Genetic diversity of lentiviruses in non-human-primates. AIDS Rev 2001 ; 3 : 3-10.
- 4. Courgnaud V, Salemi M, Pourrut X, et al. Characterization of a novel simian immunodeficiency virus with a vpu gene from greater spot-nosed monkeys (Cercopithecus nictitans) provides new insights into simian/human immunodeficiency virus phylogeny. J Virol 2002 ; 76 : 8298-309.
- 5. Souquiere S, Bibollet-Ruche F, Robertson DL, et al. Wild Mandrillus sphinx are carriers of two types of lentivirus. J Virol 2001 ; 75 : 7086-96.
- 6. Muller MC, Barre-Sinoussi F. SIVagm : genetic and biological features associated with replication. Front Biosci 2003 ; 8 : 1170-85.
- 7. Popper SJ, Sarr AD, Travers KU, et al. Lower human immunodeficiency virus (HIV) type 2 viral load reflects the difference in pathogenicity of HIV-1 and HIV-2. J Infect Dis 1999 ; 180 : 1116-21.
- 8. Hirsch VM, Johnson PR. Pathogenic diversity of simian immunodeficiency viruses. Virus Res 1994 ; 32 : 183-203.
- 9. Fultz PN, McClure HM, Anderson DC, Switzer WM. Identification and biologic characterization of an acutely lethal variant of simian immunodeficiency virus from sooty mangabeys (SIV/SMM). AIDS Res Hum Retrovir 1989 ; 5 : 397-409.
- 10. Hirsch VM, Dapolito G, Johnson PR, et al. Induction of AIDS by simian immunodeficiency virus from an African green monkey : species-specific variation in pathogenicity correlates with the extent of in vivo replication. J Virol 1995 ; 69 : 955-67.
- 11. Novembre FJ, Saucier M, Anderson DC, et al. Development of AIDS in a chimpanzee infected with human immunodeficiency virus type 1. J Virol 1997 ; 71 : 4086-91.
- 12. Kestler HW, Ringler DJ, Mori K, et al. Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell 1991 ; 65 : 651-62.
- 13. Mariani R, Kirchhoff F, Greenough TC, et al. High frequency of defective nef alleles in a long-term survivor with nonprogressive human immunodeficiency virus type 1 infection. J Virol 1996 ; 70 : 7752-64.
- 14. Novembre FJ, Johnson PR, Lewis MG, et al. Multiple viral determinants contribute to pathogenicity of the acutely lethal simian immunodeficiency virus SIVsmmPBj variant. J Virol 1993 ; 67 : 2466-74.
- 15. Tao B, Fultz PN. Pathogenicity and comparative evolution in vivo of the transitional quasispecies SIVsmmPBj8. Virology 1999 ; 259 : 166-75.
- 16. Rudensey LM, Kimata JT, Long EM, et al. Changes in the extracellular envelope glycoprotein of variants that evolve during the course of simian immunodeficiency virus SIVmne infection affect neutralizing antibody recognition, syncytium formation, and macrophage tropism but not replication, cytopathicity, or CCR-5 coreceptor recognition. J Virol 1998 ; 72 : 209-17.
- 17. Hirsch VM, Sharkey ME, Brown CR, et al. Vpx is required for dissemination and pathogenesis of SIV(SM) PBj : evidence of macrophage-dependent viral amplification. Nat Med 1998 ; 4 : 1401-8.
- 18. Courgnaud V, Laure F, Fultz PN, et al. Genetic differences accounting for evolution and pathogenicity of simian immunodeficiency virus from a sooty mangabey monkey after cross-species transmission to a pig-tailed macaque. J Virol 1992 ; 66 : 414-9.
- 19. Chakrabarti LA, Ivanovic T, Cheng-Mayer C. Properties of the surface envelope glycoprotein associated with virulence of simian-human immunodeficiency virus SHIV(SF33A) molecular clones. J Virol 2002 ; 76 : 1588-99.
- 20. Liu HY, Soda Y, Shimizu N, et al. CD4-Dependent and CD4-independent utilization of coreceptors by human immunodeficiency viruses type 2 and simian immunodeficiency viruses. Virology 2000 ; 278 : 276-88.
- 21. Overbaugh J, Bangham CR. Selection forces and constraints on retroviral sequence variation. Science 2001 ; 292 : 1106-9.
- 22. Courgnaud V, Saurin W, Villinger F, Sonigo P. Different evolution of simian immunodeficiency virus in a natural host and a new host. Virology 1998 ; 247 : 41-50.
- 23. Muller-Trutwin MC, Corbet S, Tavares MD, et al. The evolutionary rate of nonpathogenic simian immunodeficiency virus (SIVagm) is in agreement with a rapid and continuous replication in vivo. Virology 1996 ; 223 : 89-102.
- 24. Estaquier J, Monceaux V, Cumont MC, et al. Early changes in peripheral blood T cells during primary infection of Rhesus macaques with a pathogenic SIV. J Med Primatol 2000 ; 29 : 127-35.
- 25. Ploquin M, Diop O, Roques P, et al. Facteurs de non-pathogénicité de l’infection à SIVagm chez le singe vert d’Afrique. Virologie 2003 ; 7 : 1-9.
- 26. Bostik P, Wu P, Dodd GL, et al. Identification of protein kinases dysregulated in CD4(+) T cells in pathogenic versus apathogenic simian immunodeficiency virus infection. J Virol 2001 ; 75 : 11298-306.
- 27. Silvestri G, Sodora DL, Koup RA, et al. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity 2003 ; 18 : 441-52.
List of figures
Figure 1
Arbre phylogénétique des lignées de SIV (simian immunodeficiency virus).
Cet arbre a été construit à partir des séquences protéiques Pol de lentivirus de primates représentatifs des six lignées principales : SIVcpz, retrouvée chez les chimpanzés (Pan troglodytes) et associée au VIH-1 ; SIVsm, identifiée chez le mangabey enfumé (Cercocebus atys) et associée au VIH-2 ; SIVagm, présente dans les quatre espèces de singes verts (membres du genre Chlorocebus) ; SIVlhoest, du singe de l’Hoesti (Cercopithecus lhoesti), associée au SIVsun du cercopithèque à queue de soleil (Cercopithecus solatus) et au SIVmnd-1 des mandrills (Mandrillus sphinx) ; SIVsyk, du cercopithèque à diadème (Cercopithecus albogularis) ; SIVcol, décrite chez le colobe guéréza (Colobus guereza). Cet arbre a été obtenu par la méthode neighbor joining, et sa robustesse évaluée par un test de bootstrap (100 répliques). La longueur des branches est proportionnelle à la distance évolutive entre les différentes souches virales.