Abstracts
Résumé
La myopathie de Duchenne est due à l’absence d’une protéine du cytosquelette des fibres musculaires, la dystrophine. Celle-ci joue un rôle essentiel dans l’intégrité des complexes protéiques membranaires qui assurent la liaison avec la matrice extracellulaire. L’utrophine, codée au niveau du chromosome 6, présente 80 % d’homologie avec la dystrophine et est exprimée à la jonction neuromusculaire. L’utrophine peut-elle remplacer la dystrophine et corriger les anomalies structurelles et fonctionnelles de la myopathie ? Chez des souris transgéniques déficientes en dystrophine et surexprimant de grandes quantités d’utrophine, celle-ci occupe les structures fixant la dystrophine et la récupération des atteintes fonctionnelles, surtout mécaniques, peut être complète. Pour transformer cette approche expérimentale en traitement de la myopathie, les nombreuses recherches en cours visent à obtenir une surexpression d’utrophine dès l’apparition des symptômes myopathiques.
Summary
Duchenne muscle dystrophy results from the absence of dystrophin, a cytoskeletal protein of the muscle fibre. Dystrophin plays an essential role in the integrity of the membrane-associated protein complexes connected to the extracellular matrix. On chromosome 6 is located the gene of a protein presenting 80 % homology with dystrophin : utrophin, which is expressed at the neuromuscular junction. The review examines if utrophin can replace dystrophin and correct the structural and functional characteristics of the myopathy, and how the improvements can be quantitatively expressed. In transgenic mice, deficient in dystrophin, but overexpressing large quantities of utrophin, the latter is found on structures where dystrophin is normally located, histological signs of necrosis disappear and the recovery of functional disorders, specially affecting the mechanical properties of the muscle fibres, can be complete. The review examines also several ways of obtaining overexpression of utrophin in adult mdx mice, such as conditioned expression of the utrophin transgene (using a tetracycline-sensitive transactivator), transfection with viral vectors containing the utrophin cDNA (complete or truncated), actions on factor(s) controlling utrophin expression at the neuromuscular junction (heregulin, 4 N-acetylgalactosamine), and pharmacological ways of inducing expression (NO, arginine). Though partial improvements of the myopathy status have been obtained by these various approaches, they remain limited by their localized action and/or by the moderate level of utrophin expression obtained. Further researchs to overcome these limitations are urgently needed in order to transform the very promising effect of utrophin overexpression into a real treatment of Duchenne myopathy.
Article body
Myopathie de Duchenne et dystrophine
La myopathie de Duchenne (DMD) est une affection génétique grave qui touche toute la musculature. Elle résulte de mutation(s) affectant le gène de la dystrophine, localisé sur le chromosome X, et touche les garçons (1/3 500 naissances masculines). Il n’y a pas de traitement curatif, mais les traitements palliatifs, orthopédiques et respiratoires, améliorent la qualité de vie et le pronostic vital.
Dans la forme typique de la myopathie, la dystrophine est absente, alors que chez les sujets sains, cette protéine de 427 kDa est localisée sous la membrane plasmique des fibres musculaires. La plus grande partie de la molécule a une forme allongée. Du côté aminoterminal, la dystrophine se fixe aux filaments d’actine γ du cytosquelette, du coté carboxyterminal, elle s’attache à un complexe de glycoprotéines transmembranaires, les dystroglycanes. Ceux-ci interagissent avec un autre complexe transmembranaire, les sarcoglycanes, et avec la laminine qui s’attache au collagène extracellulaire [1], établissant une continuité mécanique entre le cytosquelette et la matrice extracellulaire. L’absence de dystrophine introduit une discontinuité et rend la fibre musculaire sensible au stress mécanique de l’activité physique. L’extrémité carboxyterminale interagit avec d’autres protéines [2] (Figure 1). La dystrophine assure ainsi l’édification de complexes protéiques combinant des fonctions de soutien et de signalisation, et son absence entraîne leur désagrégation.
Figure 1
Représentation schématique des interactions de nombreuses protéines avec la dystrophine.
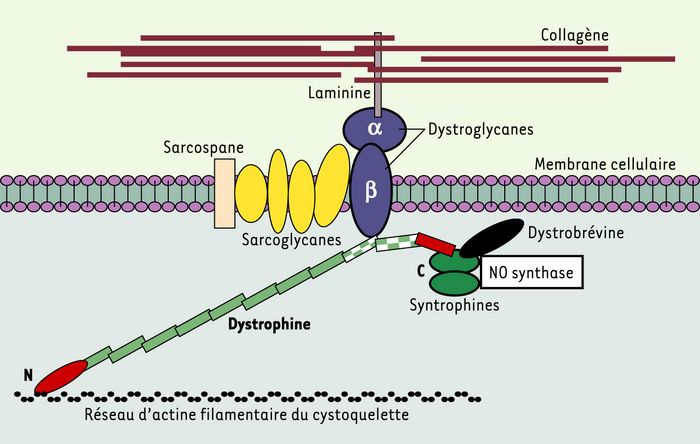
La dystrophine est une protéine de 427 kDa localisée sous la membrane plasmique des fibres musculaires (ou sarcolemme). Du côté aminoterminal (N), la dystrophine se fixe aux filaments d’actine γ constituant le cytosquelette sous-membranaire, tandis que du côté carboxyterminal, elle s’attache à un complexe de glycoprotéines transmembranaire, les dystroglycanes. Les dystroglycanes interagissent avec un autre complexe transmembranaire (formé par les sarcoglycanes et le sarcospane) et avec une forme musculaire de la laminine qui s’attache aux fibres de collagène de la matrice extracellulaire. Ainsi s’établit une continuité mécanique entre le cytosquelette d’actine et les éléments de la structure extracellulaire. L’extrémité carboxyterminale interagit avec d’autres protéines (syntrophines, dystrobrévine) qui fixent une isoforme de la NO synthase, diverses tyrosine-kinases et des canaux ioniques. L’interaction avec les dystroglycanes et les sarcoglycanes semble particulièrement importante pour assurer la résistance au stress mécanique et la liaison aux structures de la matrice extracellulaire. Le rôle fonctionnel précis de plusieurs de ces dystrophin-associated proteins reste à préciser. La localisation sous-membranaire de la NO synthase suggère un rôle paracrine du NO sur la microcirculation. Au niveau de la dystrophine, les rectangles verts représentent les séquences spectrin-like ; les différentes régions présentant des domaines bien identifiés d’interactions avec d’autres protéines sont individualisées par des couleurs et des formes différentes : domaine de liaison à l’actine (ovale allongé rouge), région de fixation aux dystroglycanes (deux rectangles à damiers vert et blanc), région de fixation aux syntrophines et dystrobrévine (rectangle rouge).
Les biopsies musculaires réalisées chez les sujets atteints de myopathie de Duchenne montrent des foyers de nécrose envahis de cellules inflammatoires et des foyers de régénération à partir de cellules souches présentes dans le muscle, les cellules satellites. La régénération, révélée par les fibres à noyaux centraux [3], compense temporairement la nécrose (Figure 2, Encadré 1). L’élévation (x 10) du taux de créatine-phosphokinase sérique (CPK) signe la fuite d’enzymes intracellulaires.
Figure 2
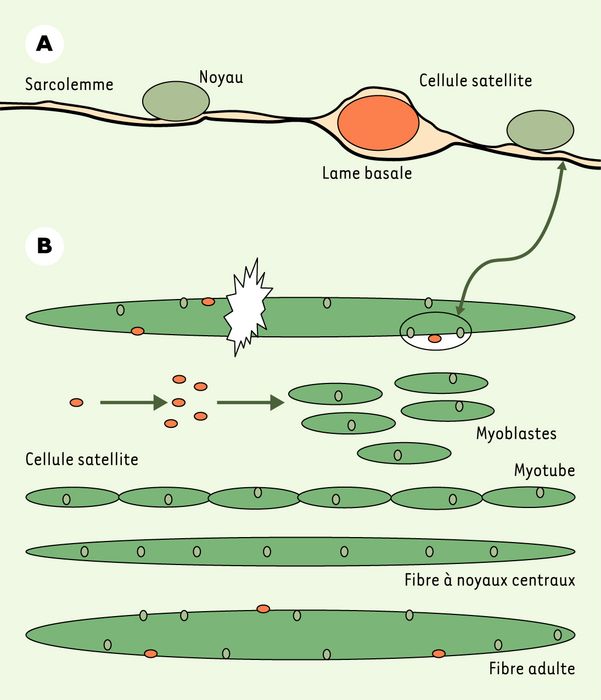
A. Localisation des noyaux des fibres musculaires et des cellules satellites associées. La fibre musculaire est un syncitium multinucléé où les noyaux occupent une position périphérique, sous-membranaire. Intimement associées à chaque fibre, mais distinctes d’elle, se trouvent les cellules satellites qui représentent 2-3 % du nombre total de noyaux. Les noyaux des cellules satellites sont situés en dehors de la membrane plasmique, leur détection se fait par immunomarquage avec des anticorps spécifiques. B. Étapes de la régénération des fibres musculaires après nécrose, à partir des cellules satellites. Lors d’une nécrose des fibres, accidentelle ou pathologique, la régénération musculaire se fait à partir des cellules satellites qui se transforment en myoblastes mononucléés et prolifératifs. Après quelques jours, ces myoblastes s’alignent bout à bout et fusionnent pour donner des myotubes polynucléés où les noyaux s’alignent en chaîne au centre du myotube. Durant la croissance en fibre adulte, les noyaux quittent cette position centrale pour se situer tous en périphérie lorsque la différenciation complète est atteinte. La fibre régénérée possède des cellules satellites.
Les répercussions fonctionnelles de l’absence de dystrophine sont multiples [4] : réduction de la force musculaire et de la résistance au stress mécanique, activité accrue de certains canaux ioniques perméables au Ca2+. La perte de l’homéostasie calcique intracellulaire est présentée comme le point de départ d’une cascade pathogénique entraînant l’activation de protéases Ca2+-dépendantes [5], mais cette hypothèse reste controversée [4]. Un déficit au niveau de la microcirculation musculaire semble aggraver la situation déjà compromise par une fibrose importante [6]. L’état dystrophique s’accompagne de variations d’expression affectant plusieurs dizaines de gènes [7-9]. Toutefois, une description claire des enchaînements pathogéniques, de leurs relations de cause à effet, et des cascades amplificatrices menant à la nécrose de la fibre fait défaut.
Il existe des modèles animaux de la myopathie de Duchenne. Les muscles des souris mdx sont ainsi dépourvus de dystrophine, mais la myopathie reste bénigne. Le chien Golden Retriever présente une myopathie très grave, modèle de la myopathie humaine, dont l’étude est rendue très difficile en raison de la sévérité particulière de l’affection.
Un analogue de la dystrophine : l’utrophine
Découverte lors de l’analyse d’ADNc de muscle foetal, l’utrophine, codée chez l’homme sur le chromosome 6, présente 80 % d’homologie avec la dystrophine [10]. Dystrophine et utrophine interagissent avec les mêmes protéines. Cette analogie entre les deux protéines explique que les premièrs préparations d’anticorps polyclonaux présentaient des réactions croisées. L’observation paradoxale d’une réaction positive à la jonction neuro-musculaire (voir ci-dessous) de muscles dépourvus de dystrophine a ainsi contribué à la découverte de l’utrophine [11].
L’utrophine est ubiquitaire. Elle est présente dans les muscles, vaisseaux, nerfs, rétine, poumons, reins et plaquettes sanguines. Dans les muscles, l’expression et la localisation de l’utrophine dépendent du degré de différenciation. Dans les myotubes et les fibres embryonnaires, l’utrophine est distribuée le long du sarcolemme. Dans le muscle adulte, elle n’est présente qu’au niveau des jonctions neuro-musculaires et myotendineuses. Dans les myopathies où se produit une régénération, l’utrophine est modérément surexprimée et localisée le long du sarcolemme [12].
À la jonction neuromusculaire, l’utrophine est colocalisée avec les récepteurs de l’acétylcholine. Sa présence dépend de facteurs de croissance d’origine présynaptique comme l’héréguline. Le rôle de l’utrophine reste mal précisé, l’inactivation de son gène chez la souris n’entraînant ni morbidité ni troubles notables de la transmission synaptique.
L’utrophine pour remplacer la dystrophine : « l’utrophinothérapie »
L’homologie structurale entre utrophine et dystrophine suggère que la première puisse remplacer la seconde. La faible surexpression spontanée d’utrophine dans la myopathie de Duchenne pourrait jouer un rôle compensateur retardant l’évolution, mais insuffisant pour éviter l’issue fatale. Chez la souris mdx, cette surexpression pourrait rendre compte du phénotype myopathique léger, car l’absence combinée de dystrophine et d’utrophine entraîne une myopathie rapidement mortelle [13, 14]. Trois circonstances favorables renforcent l’intérêt de l’utrophine comme substitut de la dystrophine : (1) le gène de l’utrophine est intact chez les patients DMD ; (2) la protéine étant exprimée, une réaction immunologique adverse n’est pas à redouter ; (3) la surexpression générale de l’utrophine chez la souris ne s’accompagne d’aucun signe pathologique. Cet article fait le point sur les approches expérimentales du traitement du déficit en dystrophine par surexpression de l’utrophine chez la souris mdx.
Approche transgénique
Le groupe de K. Davies et J. Tinsley a développé des lignées de souris mdx transgéniques surexprimant de grandes quantités d’utrophine dans les muscles [15]. Dans un premier temps, le transgène inséré codait pour une forme tronquée de l’utrophine, amputée d’environ la moitié de sa partie centrale, mais préservant les extrémités nécessaires aux interactions protéiques. Par la suite, des lignées surexprimant la molécule complète ont été obtenues. Dans les deux cas, le produit du transgène était abondant et localisé sur le pourtour et la longueur des fibres, les foyers dystrophiques typiques avaient pratiquement disparu et les complexes dystro-sarcoglycanes (DAG) avaient retrouvé leur localisation membranaire. Une étude fonctionnelle entreprise par notre groupe a porté sur les paramètres systémiques (CPK sérique), histologiques ( % de fibres à noyaux centraux), mécaniques (force musculaire maximale et résistance aux contractions avec étirements forcés, contractions « excentriques »), métaboliques (régulation du pH intracellulaire après contraction) et calciques (contenu en calcium, concentration cytosolique de calcium). Avec l’utrophine tronquée, nous avons obtenu pour tous les paramètres étudiés, des récupérations se situant dans une fourchette de 75-85 % [16, 17] (Encadré 2). Avec l’utrophine complète, la récupération atteignait 100 % pour divers paramètres de la réponse mécanique, y compris la résistance aux contractions excentriques [18]. Cette correction fonctionnelle complète a été obtenue pour la lignée Fiona où l’expression d’utrophine était 10 fois supérieure à l’expression spontanée chez la souris mdx. Des corrections incomplètes ont été obtenues pour les lignées exprimant moins d’utrophine.
Une autre façon d’obtenir une surexpression d’utrophine a été découverte fortuitement dans une construction transgénique surexprimant la N-acétylgalactosamine-transférase. Cette enzyme, localisée à la jonction neuromusculaire, assure la liaison de la 4 N-acétylgalactosamine sur une structure antigénique glycoprotéique (CT antigen) découverte dans les lymphocytes T activés, mais également présente à la jonction neuromusculaire. La surexpression de l’enzyme s’accompagne de celle du CT antigen que l’on retrouve sur tout le pourtour et la longueur des fibres musculaires. Pour des raisons encore inconnues, on observe une expression forte et généralisée d’utrophine [19]. Chez la souris mdx, cette construction transgénique induit la relocalisation des DAG et une réduction spectaculaire de l’état dystrophique [20]. Le confinement de l’utrophine à la jonction neuromusculaire peut être surmonté en agissant sur des facteurs, par exemple enzymatiques, qui y contribuent.
Chez les souris transgéniques, la surexpression de l’utrophine précède la période où apparaissent les premiers signes de l’atteinte dystrophique. Les résultats obtenus démontrent l’excellente efficacité de l’utrophine dans la prévention de la dystrophinopathie. Pour tester son efficacité dans la guérison de l’affection, il faut déclencher la surexpression de l’utrophine après l’installation du processus dystrophique. C’est le but des approches de transfection virale, de pharmacologie et de transgenèse conditionnelle.
Transfection virale
Celle-ci permet l’introduction, dans les fibres musculaires, d’un transgène inséré dans le génome d’un vecteur viral non pathogène. Idéalement, il faudrait transfecter l’ensemble de la musculature via la circulation sanguine, mais seule l’injection locale intramusculaire de la suspension virale permet d’atteindre des taux d’infections dépassant 25 % des fibres d’un muscle. Le rejet immunologique du virus est fortement réduit si l’injection est faite à des souriceaux dont le système immunitaire est immature. Dans ces conditions, une expression stable d’utrophine (> 60 jours) est obtenue dans 35 % des fibres du muscle [21]. On observe une réduction significative des foyers inflammatoires, du pourcentage de fibres centronuclées et de la sensibilité aux contractions excentriques. Cette approche a été étendue au chien dystrophique dont on sait la gravité de l’affection. Aux sites d’injection, l’expression d’utrophine est stable, dépasse nettement le niveau spontané, et s’étend à l’ensemble du sarcolemme. On note une relocalisation des DAG et une réduction nette des foyers nécrotiques et de la fibrose. Les deux approches suivantes permettent, en principe, une surexpression d’utrophine étendue à toute la musculature.
Approches pharmacologiques
Héréguline
L’expression de l’utrophine à la jonction neuromusculaire étant sous la dépendance de facteurs neurotrophiques, il était logique de les utiliser pour augmenter l’expression. Effectivement, l’injection intrapéritonéale d’héréguline à la souris mdx pendant trois mois augmente l’expression d’utrophine de 2-3 fois [22], sans signe de toxicité systémique. On observe une réduction modérée de la sensibilité aux contractions excentriques et du pourcentage de fibres à noyaux centraux.
Activation de la NO synthase
Dans les fibres musculaires, l’expression de l’utrophine est limitée à la période foetale qui est « récapitulée » lors de la régénération qui suit une dégénérescence accidentelle ou liée à une dystrophie active. La persistance de l’utrophine le long des fibres dans la DMD pourrait indiquer le maintien d’un certain état foetal. Considérant le gène de l’utrophine comme un gène foetal, Chaubourt et al. ont cherché à le « réactiver » par apport de monoxyde d’azote (NO) au moyen d’injections intrapéritonéales répétées de L-arginine, substrat de la NO synthase. Ces auteurs ont obtenu une surexpression d’utrophine s’étendant à l’ensemble du sarcolemme, chez les souris normales et dystrophiques [23]. L’imagerie fonctionnelle des muscles in situ, par résonance magnétique nucléaire, montre une texture nettement plus homogène que celle des muscles de souris mdx non traitées. La force du diaphragme est améliorée [24].
Transgenèse conditionnelle
Dans ce système, l’expression du transgène est conditionnée à son interaction avec un élément transactivateur. Le plus utilisé est une protéine se liant spécifiquement à la tétracycline. En présence de l’antibiotique, l’interaction est inhibée et le transgène reste inactif. Ce contrôle est réversible et offre la possibilité de déclencher ou de réprimer l’expression du transgène à n’importe quel moment de la vie de l’animal. Ce type de construction génétique a été développé pour l’expression conditionnelle de l’utrophine dans les muscles de souris mdx à différents moments (avant et après la naissance), suivie d’une analyse de l’état dystrophique des muscles à l’âge adulte (3 mois) [25]. Quantitativement, la surexpression par rapport à l’état mdx est modérée, bien inférieure à celle obtenue dans la lignée transgénique Fiona. Elle est cependant suffisante pour obtenir une relocalisation des DAG, une réduction du pourcentage de fibres centronucléées et une récupération de 40 % pour la résistance aux contractions excentriques [25]. Un certain niveau de guérison a donc pu être obtenu.
Conclusions
La suppléance fonctionnelle de la dystrophine par l’utrophine est aujourd’hui démontrée, lorsqu’une surexpression importante de l’utrophine est obtenue dès le stade embryonnaire. Les surexpressions stimulées après la naissance ont donné des améliorations significatives, mais l’état dystrophique persiste. On pourrait en conclure que, si elle est efficace pour prévenir la dystrophie, l’utrophinothérapie l’est beaucoup moins pour la guérir. Les approches développées pour stimuler l’expression d’utrophine chez la souris mdx adulte ont, tout au plus, permis d’augmenter celle-ci de 2 à 3 fois, nettement moins que dans la lignée transgénique Fiona, où aucun signe de dystrophie n’était plus détectable. Les résultats obtenus avec la transgenèse conditionnelle [25] indiquent que la quantité-seuil requise pour corriger un paramètre déficitaire diffère selon le paramètre considéré. La dystrophie ne pourra être vaincue que lorsque la quantité d’utrophine nécessaire à la correction du(des) paramètre(s) responsable(s) aura été obtenue.
L’utrophinothérapie vise à être un traitement causal. En cela, elle se distingue des autres approches thérapeutiques qui cherchent à contrecarrer, en aval, les conséquences pathologiques de l’absence de dystrophine. Comme le montre l’ensemble des résultats rapportés ici, cette voie est extrêmement prometteuse, mais non encore aboutie. L’effort scientifique doit porter sur : la compréhension des mécanismes liant l’absence de dystrophine et la nécrose des fibres, pour distinguer le(s) facteur(s) pathogénique(s) essentiels des symptômes associés ; la détermination du niveau d’expression d’utrophine requis pour corriger ce(s) facteur(s) ; la compréhension des mécanismes qui limitent l’expression de l’utrophine aux jonctions neuromusculaires et myotendineuses ; la découverte d’agents capables de stimuler, chez l’homme, l’expression d’utrophine au niveau requis pour corriger la dystrophie après la naissance. Ce dernier point implique le criblage à haut débit sur des modèles cellulaires pour la découverte de molécules d’intérêt qui devront être ensuite testées sur les modèles animaux, étape expérimentale incontournable. Recherches fondamentales et appliquées devront donc être menées de front.
Appendices
Références
- 1. Matsumura K, Campbell KP. Dystrophin-glycoprotein complex : its role in the molecular pathogenesis of muscular dystrophies. Muscle Nerve 1994 ; 17 : 2-15.
- 2. Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev 2002 ; 82 : 291-329.
- 3. Engel AG, Yamamoto M, Fishbeck KH. Dystrophinopathies. In : Engel AG, Franzini-Armstrong C, eds. Myology : basic and clinical. New York : McGraw-Hill, 1994 : 1133-97.
- 4. Gillis JM. Understanding dystrophinopathies : an inventory of the structural and functional consequences of the absence of dystrophin in muscles of the mdx mouse. J Muscle Res Cell Motil 1999 ; 20 : 605-25.
- 5. Turner PR, Westwood T, Regen CM, Steinhardt RA. Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature 1988 ; 355 : 735-8.
- 6. Loufrani L, Matrougui K, Gorny D, et al. Flow (shear stress)-induced endothelium-dependent dilation is altered in mice lacking the gene encoding for dystrophin. Circulation 2001 ; 103 : 864-70.
- 7. Rouger K, Le Cunff M, Steenman M, et al. Global/temporal gene expression in diaphragm and hindlimb muscles of dystrophin-deficient (mdx) mice. Am J Physiol Cell Physiol 2002 ; 283 : C773-84.
- 8. Bakay M, Zhao P, Chen J, Hoffman EP. A web-accessible complete transcriptome of normal human and DMD muscle. Neuromusc Disord 2002 ; 12 (suppl 1) : S125-41.
- 9. Boer JM, de Meijer EJ, Mank EM, et al. Expression profiling in stably regenerating skeletal muscle of dystrophin-deficient mdx mice. Neuromusc Disord 2002 ; 12 (suppl 1) : S118-24.
- 10. Tinsley JM, Blake DJ, Roche A, et al. Primary structure of dystrophin-related protein. Nature 1992 ; 360 : 591-2.
- 11. Fardeau M, Tomé FMS, Collin H, et al. Présence d’une protéine de type dystrophine au niveau de la jonction neuromusculaire dans la dystrophie musculaire de Duchenne et la souris mutante mdx. CR Acad Sci Paris Ser III 1990 ; 311 : 197-204.
- 12. Mizumo Y, Nonaka I, Hirai S, Ozawa E. Reciprocal expression of dystrophin and utrophin in muscles of Duchenne muscular dystrophy patients, female DMD carriers and control subjects. J Neurol Sci 1993 ; 119 : 43-52.
- 13. Deconinck AE, Rafael JA, Skinner JA, et al. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 1997 ; 90 : 717-27.
- 14. Deconinck N, Rafael JA, Beckers-Bleukx G, et al. Consequences of the combined deficiency in dystrophin and utrophin on the mechanical properties and myosin composition of limb and respiratory muscles of the mouse. Neuromusc Disord 1998 ; 8 : 362-70.
- 15. Tinsley JM, Potter AC, Phelps SR, et al. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature 1996 ; 384 : 349-53.
- 16. Deconinck N, Tinsley J, De Backer F, et al. Expression of truncated utrophin leads to major functional improvements in dystrophin-deficient muscles of mice. Nat Med 1997 ; 3 : 1216-21.
- 17. Goudemant JF, Deconinck N, Tinsley JM, et al. Expression of truncated utrophin improves pH recovery in exercising muscles of dystrophic mdx mice. A 31P NMR study. Neuromusc Disord 1998 ; 8 : 371-9.
- 18. Tinsley J, Deconinck N, Fisher R, et al. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat Med 1998 ; 4 : 1441-4.
- 19. Xia B, Hoyte K, Kammesheidt A, et al. Overexpression of the CT GalNAc transferase in skeletal muscle alters myofiber growth, neuromuscular structure, and laminin expression. Dev Biol 2002 ; 242 : 58-73.
- 20. Nguyen HH, Jayasinha V, Xia B, et al. Overexpression of the cytotoxic T cell GalNAc transferase in skeletal muscle inhibits muscular dystrophy in mdx mice. Proc Natl Acad Sci USA 2002 ; 99 : 5616-21.
- 21. Gilbert R, Nalbantoglu J, Petrof BJ, et al. Adenovirus-mediated utrophin gene transfer mitigates the dystrophic phenotype of mdx mouse muscles. Hum Gene Ther 1999 ; 10 : 1299-310.
- 22. Krag TOB, Juel-Jansen C, Hansen-Scwartz J, et al. Heregulin-mediated utrophin upregulation in mdx mice. J Neurol Sci 2002 ; 199 (suppl 1) : S89.
- 23. Chaubourt E, Fossier P, Baux G, et al. Nitric oxide and l-arginine cause an accumulation of utrophin at the sarcolemma : a possible compensation for dystrophin loss in Duchenne muscular dystrophy. Neurobiol Dis 1999 ; 6 : 499-507.
- 24. Delaporte S, Voisin V, Sébrié C, et al. The increase of utrophin in muscle of mdx mice treated by L-arginine reverses dystrophic phenotype and improves mechanical performance. J Neurol Sci 2002 ; 199 (suppl 1) : S88.
- 25. Squire S, Raymackers JM, Vandebrouck C, et al. Prevention of pathology in mdx mice by expression of utrophin : Analysis using an inducible transgenic expression system. Hum Mol Genet 2002 ; 11 : 3333-44.
List of figures
Figure 1
Représentation schématique des interactions de nombreuses protéines avec la dystrophine.
La dystrophine est une protéine de 427 kDa localisée sous la membrane plasmique des fibres musculaires (ou sarcolemme). Du côté aminoterminal (N), la dystrophine se fixe aux filaments d’actine γ constituant le cytosquelette sous-membranaire, tandis que du côté carboxyterminal, elle s’attache à un complexe de glycoprotéines transmembranaire, les dystroglycanes. Les dystroglycanes interagissent avec un autre complexe transmembranaire (formé par les sarcoglycanes et le sarcospane) et avec une forme musculaire de la laminine qui s’attache aux fibres de collagène de la matrice extracellulaire. Ainsi s’établit une continuité mécanique entre le cytosquelette d’actine et les éléments de la structure extracellulaire. L’extrémité carboxyterminale interagit avec d’autres protéines (syntrophines, dystrobrévine) qui fixent une isoforme de la NO synthase, diverses tyrosine-kinases et des canaux ioniques. L’interaction avec les dystroglycanes et les sarcoglycanes semble particulièrement importante pour assurer la résistance au stress mécanique et la liaison aux structures de la matrice extracellulaire. Le rôle fonctionnel précis de plusieurs de ces dystrophin-associated proteins reste à préciser. La localisation sous-membranaire de la NO synthase suggère un rôle paracrine du NO sur la microcirculation. Au niveau de la dystrophine, les rectangles verts représentent les séquences spectrin-like ; les différentes régions présentant des domaines bien identifiés d’interactions avec d’autres protéines sont individualisées par des couleurs et des formes différentes : domaine de liaison à l’actine (ovale allongé rouge), région de fixation aux dystroglycanes (deux rectangles à damiers vert et blanc), région de fixation aux syntrophines et dystrobrévine (rectangle rouge).
Figure 2
A. Localisation des noyaux des fibres musculaires et des cellules satellites associées. La fibre musculaire est un syncitium multinucléé où les noyaux occupent une position périphérique, sous-membranaire. Intimement associées à chaque fibre, mais distinctes d’elle, se trouvent les cellules satellites qui représentent 2-3 % du nombre total de noyaux. Les noyaux des cellules satellites sont situés en dehors de la membrane plasmique, leur détection se fait par immunomarquage avec des anticorps spécifiques. B. Étapes de la régénération des fibres musculaires après nécrose, à partir des cellules satellites. Lors d’une nécrose des fibres, accidentelle ou pathologique, la régénération musculaire se fait à partir des cellules satellites qui se transforment en myoblastes mononucléés et prolifératifs. Après quelques jours, ces myoblastes s’alignent bout à bout et fusionnent pour donner des myotubes polynucléés où les noyaux s’alignent en chaîne au centre du myotube. Durant la croissance en fibre adulte, les noyaux quittent cette position centrale pour se situer tous en périphérie lorsque la différenciation complète est atteinte. La fibre régénérée possède des cellules satellites.