Abstracts
Résumé
Le cancer gastrique est observé partout dans le monde. Sa fréquence est particulièrement élevée au Japon et en Amérique du Sud. Son incidence n’a cessé de décroître en France depuis la fin de la Seconde Guerre mondiale et, actuellement, ce cancer n’est responsable que d’environ 10 % des décès. L’infection par Helicobacter pylori, la spécificité génétique et le régime alimentaire de l’hôte sont les principaux facteurs impliqués dans ce cancer. Éradiquer Helicobacter pylori chez les apparentés au premier degré d’un sujet atteint de cancer gastrique et chez tout sujet ayant une gastrite atrophiante et un autre cancer digestif paraît une mesure préventive sage.
Summary
Gastric cancer is a worldwide cancer especially frequent in Japan and South America. This cancer affects 10 to 70 people per 100 000 according to the countries. Since the end of the Second World War, the incidence of gastric cancer has been decreasing in France and accounts for less than 10 % of mortality. Helicobacter pylori infection, host genetic background, food regimen are known to be involved in this cancer. Helicobacter pylori should be eradicated in selected patients, such as patients’ relatives with documented gastric cancer as well as patients having another gastrointestinal cancer.
Article body
Le cancer gastrique reste un problème de santé publique dans le monde, notamment au Japon, en Chine et en Amérique du Sud. Il touche de 10 à 70 habitants/100 000 par an selon les pays. En France, son incidence est de 10 à 15/100 000 [1]. Sur le plan histologique, on distingue les cancers superficiels des cancers invasifs, ces derniers pouvant être des adénocarcinomes de type intestinal bien différenciés ou de type diffus indifférencié. Le cancer gastrique est une affection multifactorielle. Helicobacter pylori (H. pylori) occupe la première place parmi les paramètres étiopathogéniques. L’infection à H. pylori provoque une gastrite qui, après plusieurs années d’évolution chronique, peut aboutir dans certains cas au cancer.
Historique
Le frère de Napoléon Bonaparte, ses deux soeurs, son père et probablement sa tante paternelle, sont tous décédés d’un cancer gastrique. Après le décès de l’empereur en exil, le communiqué officiel faisait état d’une perforation sur tumeur maligne gastrique. Malgré la rumeur publique évoquant un empoisonnement, le communiqué officiel avait beaucoup de chance de convaincre tant l’arbre généalogique des Bonaparte pouvait suggérer une forme familiale de cette maladie. Cette anecdote résume les concepts étiopathogéniques dans le domaine du cancer gastrique puisque cet arbre généalogique fut ensuite cité pendant longtemps comme preuve d’une prédisposition génétique à ce cancer.
Au xixe siècle, le cancer gastrique constituait à lui seul 40 % des cancers chez l’homme [2]. Dans la première moitié du xxe siècle, l’incidence de ce cancer était élevée [3]. En 1927, la théorie « héréditaire », d’abord fortement soutenue, fut reléguée au second plan avec la description, pour la première fois, d’une gastrite chronique pouvant correspondre à un état de prédisposition au cancer gastrique [3]. La baisse de l’incidence du cancer gastrique dans les pays industrialisés (exception faite du Japon), durant la deuxième moitié du xxe siècle, correspond à une amélioration du niveau de vie et de l’hygiène. La découverte de H. pylori au début des années 1980 fait tomber le dogme du cancer héréditaire en donnant naissance au concept de cancer d’origine « infectieuse » et permet d’établir une séquence physiopathologique : l’infection par H. pylori acquise souvent dans l’enfance, avant l’âge de 10 ans [4], provoque l’apparition d’une gastrite aiguë interstitielle ; celle-ci fait ensuite place à une gastrite chronique qui reste le plus souvent asymptomatique tout en évoluant lentement. Cette gastrite peut s’associer dans certains cas à une forme ulcéreuse gastrique ou duodénale, au lymphome de type MALT (mucosa-associated lymphoid tissue) ou au cancer.
Données épidémiologiques
L’infection à H. pylori touche plus de 50 % de la population dans le monde. Sa prévalence varie selon le lieu géographique, le statut socioéconomique et l’âge : 22 % à l’âge de 20 ans et 66 % à l’âge de 60 ans dans les pays développés [5]. H. pylori est immunogène, mais la réponse immunitaire ne protège pas contre les maladies qu’il induit.
La survenue d’un cancer gastrique invasif toucherait seulement 3 % des sujets porteurs de H. pylori [6]. La relation entre l’infection à H. pylori et l’adénocarcinome gastrique est suggérée avant tout par des données épidémiologiques. Ainsi, J. Parsonnet et al. [7], dans une étude cas-témoin, ont sélectionné 109 malades atteints de cancer gastrique et 109 sujets témoins appariés sur l’âge, le sexe, l’ethnie et l’origine géographique. Les individus de l’étude étaient issus d’une cohorte de 128 992 sujets pour lesquels du sérum était disponible pour une détection d’anticorps anti-H. pylori (le délai moyen entre la date du recueil du sérum et le diagnostic du cancer gastrique était de 14,2 années). La sérologie de H. pylori était positive chez 84 % des malades et 61 % des témoins. Dans cette étude, l’odds ratio (intervalle de confiance à 95 %) de cancer gastrique chez les patients infectés par H. pylori était de 3,6 (1,8-7,3). La récente méta-analyse du Groupe coopératif d’étude de cancer et Helicobacter [8] qui regroupe 12 études, soit 1 228 sujets atteints d’un cancer gastrique et 3 406 témoins, confirme cette conclusion (Figure 1). C’est sur ces données épidémiologiques, que, en 1994, l’Agence internationale de recherche sur le cancer a classé H. pylori parmi les carcinogènes de classe I, c’est-à-dire jouant un rôle carcinogène certain chez l’homme [9].
Figure 1
Études épidémiologiques montrant la potentialisation du risque de cancer gastrique par H. pylori.
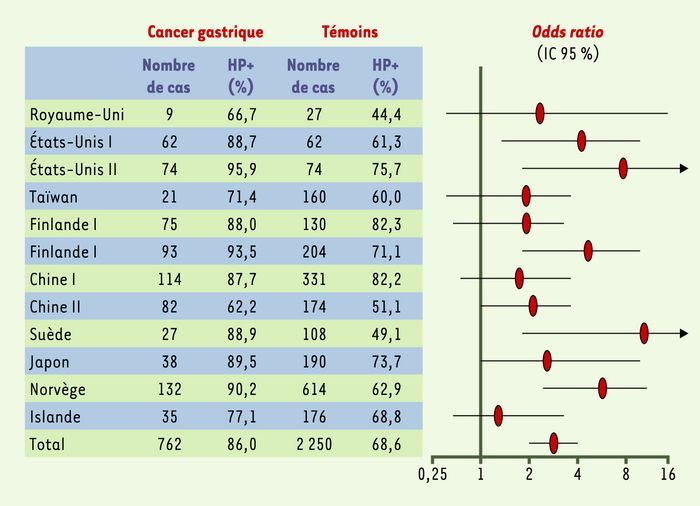
Résultats de 12 études prospectives réalisées dans 9 pays montrant les odds ratio d’association entre infection par H. pylori et cancer gastrique. La combinaison des résultats permet d’obtenir un odds ratio global de l’ordre de 3 pour l’association entre H. pylori et cancer gastrique. HP+(%) : pourcentage de sujets positifs pour H. pylori dans chaque étude ; IC 95 % : intervalle de confiance à 95 % (d’après [8]).
Lésions précancéreuses et H. pylori
L’homme est le réservoir de H. pylori et la transmission se fait vraisemblablement par les selles ou la salive d’un sujet infecté [4]. Après ingestion de la bactérie, l’estomac est le premier site où H. pylori se niche, créant différentes formes de gastrite qui n’aboutissent pas toutes au cancer. La gastrite limitée à l’antre (partie horizontale de l’estomac) est généralement associée à une maladie ulcéreuse et une hypersécrétion acide, et ne s’associe jamais à un cancer gastrique [10, 11]. À l’inverse, la gastrite du corps (partie verticale de l’estomac), souvent isolée, s’accompagne progressivement d’une baisse de la sécrétion acide due à une raréfaction des cellules pariétales. C’est dans cette situation qu’une atrophie totale peut s’installer au bout de plusieurs années. La filiation entre gastrite chronique atrophique et cancer a été établie par P. Correa bien avant la découverte de H. pylori [11]. Au Japon, le risque relatif de survenue du cancer gastrique compliquant une gastrite atrophique est multiplié par 4 en présence de H. pylori, et celui d’un cancer gastrique superficiel est multiplié par 5 [12]. Les cancers de type diffus plus que d’autres formes de cancer sont associés à H. pylori : le risque relatif d’un cancer diffus est de 8, celui d’une forme différenciée intestinale de 3 à 4 [7].
La métaplasie intestinale, véritable lésion précancéreuse, est plus fréquemment retrouvée chez les sujets infectés par H. pylori. Elle est irréversible, même après l’éradication de H. pylori. La bactérie peut disparaître spontanément au stade de métaplasie intestinale et la concentration sérique d’anticorps anti-H. pylori diminuer, voire disparaître. Toutefois, la transformation maligne favorisée par H. pylori est loin d’être une évolution fréquente, eu égard à l’incidence importante de la gastrite chronique et à la fréquence de métaplasies intestinales dans la population générale. Pour expliquer ces paradoxes, il faut admettre que le schéma classique de carcinogenèse proposé par P. Correa [11] n’est pas valable pour tous les cancers.
Facteurs augmentant le risque carcinogène de H. pylori
Le cancer gastrique est une maladie multifactorielle. Si la bactérie représente un maillon important dans sa genèse, le type de la souche bactérienne, mais aussi le régime alimentaire et surtout la spécificité génétique de l’hôte sont également des facteurs qui participent au développement de ce cancer (Figure 2).
Figure 2
Schéma physiopathogénique de l’implication de H. pylori dans le cancer gastrique.
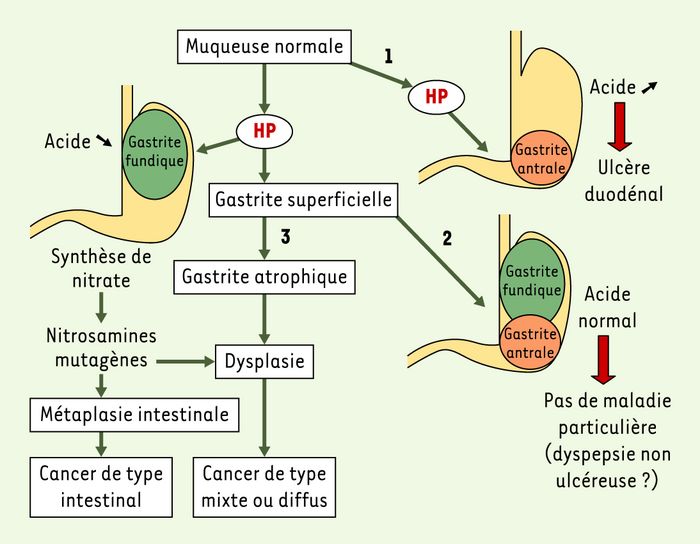
H. pylori (HP) peut entraîner une gastrite à prédominance antrale (voie 1) ; c’est une forme associée à une hypersécrétion acide et à la maladie ulcéreuse. Il peut entraîner une gastrite mixte (touchant l’antre et le fundus), la sécrétion acide n’est pas modifiée et aucune maladie particulière (à l’exception de la dyspepsie non ulcéreuse) n’y est associée (voie 2) : c’est la forme la plus fréquente. Dans certains cas, la gastrite est à prédominance fundique, la sécrétion acide est diminuée et l’atrophie s’installe (voie 3). Cette forme est volontiers associée au cancer en passant par des étapes précancéreuses. Deux hypothèses sont actuellement retenues pour expliquer la diversité des lésions précancéreuses. Dans l’hypothèse proposée par P. Correa [11], H. pylori semble altérer les propriétés physiques et chimiques du mucus gastrique, le rendant plus sensible aux facteurs carcinogènes. Le régime alimentaire (richesse en sel et pauvreté en acide ascorbique) de l’hôte influence le processus carcinogène. La hausse du pH favoriserait ainsi une prolifération de la flore bactérienne intestinale douée d’une activité nitrate réductase, qui métabolise les nitrates en produisant de la nitrosamine, substance cancérigène [32]. Une muqueuse de type intestinal remplace progressivement la muqueuse gastrique, c’est la métaplasie gastrique. Le cancer qui va ainsi se développer est plutôt de type intestinal : c’est la forme la plus fréquente des cancers gastriques. Il est à noter que le cancer de type diffus survient plutôt chez le sujet jeune [10]. La spécificité génétique de l’hôte, en particulier lors de la réponse inflammatoire, est un des facteurs influençant le risque de survenue du cancer [25-29].
Rôle de la souche bactérienne
Ces dernières années, de nombreux travaux se sont intéressés à la caractérisation des facteurs de virulence de H. pylori et à leurs implications pathogéniques. Trois protéines, CagA, vacA et HP-MP1 ont ainsi été caractérisées.
La protéine CagA (cytotoxin-associated genes A), de masse moléculaire 128 kDa, est exprimée dans 60 % à 70 % des souches de H. pylori, avec des variations selon les zones géographiques. Cette protéine est codée par un gène appartenant à l’îlot de pathogénicité Cag. Plusieurs protéines ayant de fortes homologies avec les protéines constituant les appareils bactériens de sécrétion de type IV ((→) m/s 2002, n° 4, p. 439) sont codées par l’îlot Cag. Lorsqu’il est fonctionnel, l’appareil de sécrétion permet l’induction de la synthèse d’IL-8 (interleukine-8) à partir des cellules épithéliales. H. pylori active cette production par deux voies. La première inclut le facteur nucléaire NF-κB via l’activation de la p21-kinase et la seconde l’activation de la protéine AP-1 (activation protein-1) via les petites GTPases de la famille Rho [6]. Des études cliniques révèlent que la présence de la protéine CagA est corrélée à l’évolutivité de la gastrite, à l’atrophie gastrique et au cancer [12, 13]. Ces résultats ne sont pas retrouvés par tous les auteurs [14, 15], mais une activation différentielle des voies de signalisation dans les cellules de l’hôte selon le contenu génétique des souches associées à des pathologies gastriques différentes a déjà été rapportée [16]. La majorité des gènes induits lors d’une infection par H. pylori associée à un cancer gastrique codent pour des facteurs transcriptionnels (C-jun, BTEB2/KLF5 [kruppel-like factor 5 intestinal], ETR101) ou des facteurs impliqués dans la voie de transduction du signal (MAP-kinases, insulin growth factor) [17].
Bien que le gène vacA soit présent dans tous les isolats cliniques, la protéine VacA, une cytotoxine vacuolisante active, est produite dans 50 % à 65 % des souches de H. pylori. Elle induit la vacuolisation des cellules épithéliales in vitro et in vivo chez l’homme. La forme allélique vacA-s1 semble associée à un risque accru d’ulcère et de cancer gastrique [6, 18]. La toxine VacA induit la formation de pores dans la membrane cellulaire. Ces pores possèdent une sélectivité pour les anions et seraient responsables de la constitution de larges vacuoles via l’augmentation de l’activité de la V-ATPase, une pompe à protons électrogénique [18]. Le rôle précis de cette protéine dans la carcinogenèse est encore mal connu.
Une protéine membranaire de H. pylori de 16 kDa, appelée HP-MP1, est pro-inflammatoire et stimule en particulier la production de TNFα (tumor necrosis factor α), IL-1, IL-8 et MIP1α (mitogen inhibitor peptide α). Son potentiel carcinogène a récemment été suggéré in vitro et in vivo chez la souris [19].
Rôle de l’hypochlorhydrie
Parmi les facteurs liés à l’hôte, la sécrétion acide joue un rôle essentiel. On peut schématiquement distinguer deux situations : en cas de sécrétion acide augmentée, une gastrite antrale se développe préférentiellement, le risque d’ulcère duodénal est majoré et celui de cancer réduit ou nul. En revanche, une sécrétion acide basse favorise le développement d’une pangastrite et augmente le risque de cancer gastrique [20]. Dans ce cas, l’hypergastrinémie réactionnelle jouerait un rôle essentiel dans la croissance tumorale par son effet trophique. Dans le modèle de cancer gastrique de gerbille du Caucase, l’hypergastrinémie induite par H. pylori a pour conséquence une augmentation de la prolifération cellulaire épithéliale antrale en l’absence de gastrite aiguë. En présence d’une telle gastrite, l’augmentation de la prolifération est compensée par une stimulation de l’apoptose [21]. Dans un modèle de souris génétiquement modifiée (INS-GAS) présentant initialement une hypergastrinémie, puis une atrophie gastrique et une hypochlorhydrie, l’infection par Helicobacter felis accélère le développement d’un cancer gastrique [22].
Rôle de la susceptibilité génétique de l’hôte
En 1998, le concept de cancer gastrique héréditaire s’est concrétisé pour la première fois sur le plan moléculaire par la mise en évidence de mutations germinales du gène CDH1/E-cadhérine dans trois familles, mutations décrites par la suite dans des familles d’origines ethniques différentes [23]. Bien que des mutations somatiques du même gène aient pu être identifiées dans de nombreux cas sporadiques, leur prévalence reste faible [24]. Il est intéressant de noter que la prévalence de H. pylori, estimée sur la sérologie, est plus importante chez les sujets avec (69 %) que chez les sujets sans (44 %) antécédent familial de cancer gastrique [24]. L’atrophie gastrique et l’hypochlorhydrie sont plus fréquentes (27 %) chez les sujets apparentés au premier degré de patients atteints d’un cancer de l’estomac que chez les témoins (3 %), malgré une incidence similaire d’infection à H. pylori (63 % versus 64 %) [25]. D’où l’idée qu’une susceptibilité génétique au cancer se surajoute au rôle de H. pylori. Une association entre polymorphisme du gène codant pour l’interleukine-1β (IL-1β) et risque d’atrophie gastrique et de cancer a été pour la première fois suggérée par E.M. El-Omar et al. [26, 27] et confirmée par d’autres auteurs [28] (Tableau I). Les porteurs de l’allèle 511T du gène codant pour l’IL-1β ont une sécrétion acide plus faible que ceux porteurs de l’allèle 511C, ce qui suggère que le polymorphisme du gène codant pour l’IL-1β conditionne le niveau de la sécrétion acide lorsqu’un processus inflammatoire chronique est présent au niveau de la muqueuse gastrique [29]. Chez la souris, une association étroite a été trouvée entre la réponse cytokinique de type Th1 (interféron γ, IL-1, TNFα), le développement d’une atrophie et la survenue de lésions prénéoplasiques. Inversement, la conversion d’une réponse Th1 en Th2 protège ces animaux contre l’atrophie [30].
Tableau I
Polymorphisme génétique et risque de cancer gastrique.

L’équipe d’E.M. El-Omar [29-31] a analysé le risque de cancer gastrique en fonction du polymorphisme génique de l’hôte. Dans trois populations différentes, anglaise, américaine et polonaise, les risques de survenue d’un adénocarcinome gastrique (odds ratio, intervalle de confiance à 95 %) sont établis en fonction des cytokines étudiées : IL-1 (interleukine-1), TNF (tumor necrosis factor) et IL-10. (les données de ce tableau ont été présentées au congrès annuel de la Société américaine de gastroentérologie, Digestive disease week, San Francisco, mai 2002).
Les gènes codant pour des peptides régulateurs de la prolifération cellulaire comme les peptides en « trèfle » (trefoil factor family) sont un domaine d’étude intéressant pour définir une spécificité d’hôte. Les souris invalidées pour ce gène développent des adénomes et des cancers et, chez l’homme, il existe une perte d’expression de cette famille de peptides dans 44 % des cancers gastriques [31].
L’éradication de H. pylori permet-elle de prévenir l’adénocarcinome gastrique ?
L’étude de N. Uemura et al. [10] apporte une réponse partielle à cette question. Ces auteurs ont assuré un suivi endoscopique de 1 526 patients, pendant une durée moyenne de 7,2 + 2,5 ans. Un cancer gastrique a été détecté chez 36 (2,9 %) des patients infectés par H. pylori, mais chez aucun des patients non infectés. Parmi les patients positifs pour H. pylori, le risque de survenue de cancer était plus élevé, sauf chez ceux ayant fait un ulcère duodénal. Cette étude démontre pour la première fois le risque réel de laisser évoluer une gastrite « H. pylori positif ». Une autre étude japonaise montre le risque élevé de récidive de cancer chez les sujets ayant subi un traitement conservateur lors de la découverte d’un premier cancer superficiel : l’incidence des récidives de cancer gastrique superficiel dans les quatre ans était de 0 % chez les patients ayant bénéficié d’une éradication de H. pylori, contre 9 % chez les autres [32].
Malgré ces données, il n’existe pas à l’heure actuelle de consensus sur l’éradication systématique de H. pylori pour diminuer le risque de cancer gastrique, en raison de la faible incidence de ce type de cancer dans notre pays, de la variabilité des souches impliquées, du caractère multifactoriel de la survenue du cancer et du coût d’une éradication de H. pylori à l’échelle d’une population. En supposant que l’éradication prévienne 30 % des cancers gastriques, le rapport coût/efficacité d’un dépistage suivi d’une éradication systématique de H. pylori s’élèverait à 25 000 $US par année de vie sauvée, ce chiffre étant variable selon la prévalence du cancer dans la population étudiée : de 4 500 $US pour les Américains d’origine japonaise à 35 000 $US pour les Caucasiens [33]. Toutes ces données incitent donc à une politique d’éradication ciblée de H. pylori.
Les sujets à cibler pour l’éradication de l’infection à H. pylori appartiennent à des populations à risque : patients après traitement d’un cancer gastrique superficiel ou sujets ayant des antécédents familiaux de cancer gastrique, comme cela a été défini par la conférence de consensus du Groupe européen d’étude de H. pylori en 1996. Ces indications pourraient s’étendre aux groupes ethniques à risque. En attendant, il est prudent de proposer l’éradication de H. pylori chez tous les parents au premier degré de patients atteints de cancer gastrique et chez tous les patients présentant une gastrite atrophiante ou un foyer de métaplasie intestinale.
Conclusions
Le cancer gastrique fait maintenant partie des cancers dans la genèse desquels un agent microbien est identifié. La baisse de l’incidence de ce cancer dans la plupart des pays industrialisés peut s’expliquer en partie par la diminution de la prévalence d’infections par H. pylori. Il est toutefois difficile de proposer à l’heure actuelle une éradication de cette bactérie à l’échelle des populations dans l’espoir de voir disparaître ce cancer. Des études devraient être encore menées pour permettre d’identifier les sujets infectés à risque (caractérisation des souches bactériennes impliquées dans le cancer gastrique et marqueurs de susceptibilité prédictifs) devant bénéficier d’une cure d’éradication pour les mettre à l’abri de la survenue d’un cancer gastrique.
Appendices
Références
- 1. Launoy G, Grosclaude P, Pienkowski P, et al. Cancers digestifs en France. Comparaison de l’incidence dans 7 départements et estimation de l’incidence pour la France entière. Gastroenterol Clin Biol 1992 ; 16 : 633-8.
- 2. Osler W, Mc Crae T. Cancer of the stomach. Philadelphia : Blakiston’s son and Co, 1900 : 157 p.
- 3. Parsonnet J. When heredity is infectious. Gastroenterology 2000 ; 118 : 222-7.
- 4. Malaty HM, El-Kasabany A, Graham D, et al. Age at acquisition of Helicobacter pylori infection : a follow-up study from infancy to adulthood. Lancet 2002 ; 359 : 931-5.
- 5. Gasbarrini G, Pretolani S, Bonvicini F, et al. A population-based study of Helicobacter pylori infection in a european country : the San Marino study. Relation with gastrointestinal diseases. Gut 1995 ; 36 : 838-44.
- 6. Peek RM, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer 2002 ; 2 : 28-37.
- 7. Parsonnet J, Friedman GD, Vandersteen DP, et al.Helicobacter pylori infection and the risk of gastric cancer. N Engl J Med 1991 ; 325 : 1127-31.
- 8. Helicobacter and cancer cooperative group. Gastric cancer and Helicobacter pylori : a combined analysis of 12 case control studies nested within prospective cohorts. Gut 2001 ; 49 : 347-53.
- 9. International agency for research of cancer (IARC). Infectionwith Helicobacter pylori. Monographs on the evaluation of carcinogenic risks to humans, vol. 61. Lyon : IARC, 1994 : 177-240.
- 10. Uemura N, Okamoto S, Yamamoto S, et al. Helicobacter pylori infection and development of gastric cancer. N Engl J Med 2001 ; 345 : 784-9.
- 11. Correa P. Human gastric carcinogenesis : a multistep and multifactorial process. First american cancer society award lecture on cancer epidemiology and prevention. Cancer Res 1992 ; 52 : 6735-40.
- 12. Parsonnet J, Friedman GD, Orentreich N, Vogelman H. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut 1997 ; 40 : 297-301.
- 13. Labenz J, Malfertheiner P. Helicobacter pylori in gastro-oesophageal reflux disease : causal agent, independent or protective factor ? Gut 1997 ; 41 : 277-80.
- 14. Van der Hulst RWM, Van der Ende A, Dekker FW, et al. Effect of Helicobacter pylori eradication on gastritis in relation to cagA : a prospective one-year follow-up study. Gastroenterology 1997 ; 40 : 297-301.
- 15. Mitchell HM, Hazell SL, Li YY, Hu PJ. Serological response to specific Helicobacter pylori antigens : antibody against CagA antigen is not predictive of gastric cancer in a developing country. Am J Gastroenterol 1996 ; 91 : 1785-8.
- 16. Israel DA, Salama N, Arnold CN, et al. Helocobacter pylori strain-specific differences in genetic content, identified by microarray, influence host inflammatory responses. J Clin Invest 2001; 107 : 611-20.
- 17. Chiou GC, Chan CC, Sheu DL, et al. Helicobacter pylori infection induced alteration of gene expression in human gastric cells. Gut 2001 ; 48 : 598-604.
- 18. Galmiche A, Rassow J, Doye A, et al. The N-terminal 34 kDa fragments of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrom C release. EMBO J 2000 ; 68 : 6361-70.
- 19. Suganuma M, Kurusu M, Okabe S, et al. Helicobacter pylori membrane protein 1 : a new carcinogenic factor of Helicobacter pylori. Cancer Res 2001 ; 61 : 6356-9.
- 20. Hansson LE, Nyrén O, Hsing AW, et al. The risk of stomach cancer in patients with gastric or duodenal ulcer disease. N Engl J Med 1996 ; 335 : 242-9.
- 21. Peek RM, Wirth HP, Moss SF, et al. Helicobacter pylori alters gastric epithelial cell cycle events and gastrin secretion in Mongolian gerbils. Gastroenterology 2000 ; 118 : 48-59.
- 22. Wang TC, Dangler CA, Chen D, et al. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology 2000 ; 118 : 36-47.
- 23. Caldas C, Carneiro F, Lynch HT, et al. Familial gastric cancer : overview and guidelines for management. J Med Genet 1999 ; 36 : 873-80.
- 24. Brenner H, Bode G, Boeing H. Helicobacter pylori among offspring of patients with stomach cancer. Gastroenterology 2000 ; 118 : 31-5.
- 25. El-Omar EM, Oien K, Murray LS, et al. Increased prevalence of precancerous changes in relatives of gastric cancer patients : critical role of H. pylori. Gastroenterology 2000 ; 118 : 22-30.
- 26. El-Omar EM, Carrington M, Chow WH. IL1 polymorphisms associated with increased risk of gastric cancer. Nature 2000 ; 404 : 398-402.
- 27. El-Omar EM. The importance of interleukin 1β in Helicobacter pylori associated disease. Gut 2001 ; 48 : 743-47.
- 28. Machado JC, Pharoah P, Sousa S, et al. Interleukin-1β and interleukin-1RN polymorphisms are associated with increased risk of gastric carcinoma. Gastroenterology 2001 ; 121 : 823-29.
- 29. Hwang I, Kodama T, Kikuchi S, et al. Effect of interleukin-1 polymorphisms on gastric acid mucosal interleukin-1β production in Helicobacter pylori infection. Gastroenterology 2002 ; 123 : 1793-803.
- 30. Fox JG, Beck P, Dangler CA, et al. Concurrent eneric helminth infection modulates inflammation and gastric immune responses and reduces Helicobacter-induced gastric atrophy. Nat Med 2000 ; 6 : 536-42.
- 31. Park WS, Oh RR, Park JY, et al. Somatic mutations of the trefoil factor family 1 gene in gastric cancer. Gastroenterology 2000 ; 119 : 691-8.
- 32. Uemura N, Mukai T, Okamoto S, et al. Effect of Helicobacter pylori eradication on subsequent development of cancer after endoscopic resection of early gastric cancer. Cancer Epidemiol Biomarkers 1997 ; 6 : 639-42.
- 33. Parsonnet J, Harris RA, Hack HM, Owens DK. Modeling cost effectiveness of Helicobacter pylori sreening to prevent gastric cancer : a mandate for clinical trials. Lancet 1996 ; 348 : 150-4.
List of figures
Figure 1
Études épidémiologiques montrant la potentialisation du risque de cancer gastrique par H. pylori.
Résultats de 12 études prospectives réalisées dans 9 pays montrant les odds ratio d’association entre infection par H. pylori et cancer gastrique. La combinaison des résultats permet d’obtenir un odds ratio global de l’ordre de 3 pour l’association entre H. pylori et cancer gastrique. HP+(%) : pourcentage de sujets positifs pour H. pylori dans chaque étude ; IC 95 % : intervalle de confiance à 95 % (d’après [8]).
Figure 2
Schéma physiopathogénique de l’implication de H. pylori dans le cancer gastrique.
H. pylori (HP) peut entraîner une gastrite à prédominance antrale (voie 1) ; c’est une forme associée à une hypersécrétion acide et à la maladie ulcéreuse. Il peut entraîner une gastrite mixte (touchant l’antre et le fundus), la sécrétion acide n’est pas modifiée et aucune maladie particulière (à l’exception de la dyspepsie non ulcéreuse) n’y est associée (voie 2) : c’est la forme la plus fréquente. Dans certains cas, la gastrite est à prédominance fundique, la sécrétion acide est diminuée et l’atrophie s’installe (voie 3). Cette forme est volontiers associée au cancer en passant par des étapes précancéreuses. Deux hypothèses sont actuellement retenues pour expliquer la diversité des lésions précancéreuses. Dans l’hypothèse proposée par P. Correa [11], H. pylori semble altérer les propriétés physiques et chimiques du mucus gastrique, le rendant plus sensible aux facteurs carcinogènes. Le régime alimentaire (richesse en sel et pauvreté en acide ascorbique) de l’hôte influence le processus carcinogène. La hausse du pH favoriserait ainsi une prolifération de la flore bactérienne intestinale douée d’une activité nitrate réductase, qui métabolise les nitrates en produisant de la nitrosamine, substance cancérigène [32]. Une muqueuse de type intestinal remplace progressivement la muqueuse gastrique, c’est la métaplasie gastrique. Le cancer qui va ainsi se développer est plutôt de type intestinal : c’est la forme la plus fréquente des cancers gastriques. Il est à noter que le cancer de type diffus survient plutôt chez le sujet jeune [10]. La spécificité génétique de l’hôte, en particulier lors de la réponse inflammatoire, est un des facteurs influençant le risque de survenue du cancer [25-29].
List of tables
Tableau I
Polymorphisme génétique et risque de cancer gastrique.
L’équipe d’E.M. El-Omar [29-31] a analysé le risque de cancer gastrique en fonction du polymorphisme génique de l’hôte. Dans trois populations différentes, anglaise, américaine et polonaise, les risques de survenue d’un adénocarcinome gastrique (odds ratio, intervalle de confiance à 95 %) sont établis en fonction des cytokines étudiées : IL-1 (interleukine-1), TNF (tumor necrosis factor) et IL-10. (les données de ce tableau ont été présentées au congrès annuel de la Société américaine de gastroentérologie, Digestive disease week, San Francisco, mai 2002).