Article body
Les facteurs d’échange nucléotidiques (GEF) sont des composants obligatoires des cascades de signalisation contrôlées par les petites protéines G de la famille de l’oncogène p21Ras (appelées protéines G ci-dessous) (pour revue, voir [1]). Ces protéines régulatrices ont pour fonction biochimique de stimuler la dissociation du nucléotide GDP qui est fortement associé à la protéine G inactive ; cela favorise l’association à la protéine G du nucléotide GTP et la formation de la conformation active de la protéine G (Figure 1, gauche). Dans les cellules, ce sont les facteurs d’échange qui collectent les signaux qui déclenchent l’activation des protéines G. Pour cela, les GEF ont en général une structure modulaire dans laquelle le domaine catalytique responsable de la fonction d’échange est associé « en tandem » avec des domaines régulateurs qui déterminent les conditions à réunir pour la mise en place de la cascade. De nombreux indices s’accumulent en faveur d’un rôle des GEF en aval dans la sélection des effecteurs qui sont recrutés par la protéine G activée, une même protéine G induisant des réponses distinctes selon le GEF qui l’active (pour revue, voir [2]).
Figure 1
Le cycle GDP/GTP des protéines G.
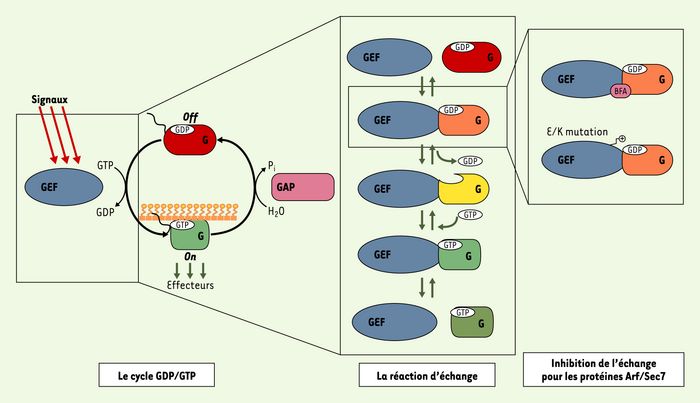
À gauche : les protéines G sont activées par l’échange du GDP par du GTP, qui est stimulé par les GEF à la réception de signaux d’activation en amont. Elles transmettent alors un message d’action en interagissant avec les partenaires spécifiques de leur conformation liée au GTP, appelés collectivement « effecteurs ». Le cycle est terminé par les « GAP », qui stimulent l’hydrolyse du GTP en GDP. Dans certains cas comme celui des protéines Arf et Rho, le cycle GDP/GTP est couplé à une translocation de la protéine G du cytosol à la membrane. Au milieu : la réaction biochimique de la dissociation du GDP stimulée par les GEF. Les complexes ternaires protéine G/nucléotide/GEF ont une affinité beaucoup plus faible que le complexe sans nucléotide. À droite : l’activation de la protéine G Arf par ses GEF à domaine Sec7 peut être inhibée par stabilisation de complexes abortifs Arf-GDP-GEF en présence de la toxine bréfeldine A ou d’une mutation ponctuelle du GEF.
Ces caractéristiques font des GEF des cibles thérapeutiques potentielles pour interrompre spécifiquement des mécanismes moléculaires impliquant une (sur)activation des protéines G dans des maladies humaines. En effet, une participation des protéines G, défectueuses ou non, à de nombreux contextes pathologiques est reconnue depuis longtemps. Le ciblage thérapeutique de ces protéines se heurte cependant à la polyvalence de leurs interactions, puisqu’elles peuvent être activées par de multiples GEF, et activer à leur tour plusieurs effecteurs. Cette caractéristique s’applique aussi à leurs régulateurs et effecteurs, qui peuvent être très spécifiques d’une protéine G donnée ou, au contraire, interagir avec plusieurs protéines G. Les stratégies d’inhibition compétitive - dans lesquelles une molécule pharmacologique empêche la protéine d’intérêt d’interagir avec ses partenaires cellulaires - interrompent ainsi l’ensemble de ses fonctions, entraînant des effets potentiellement indésirables et dangereux.
Mécanismes structuraux de l’inhibition d’un facteur d’échange nucléotidique
Nous avons décrit récemment les bases structurales d’un nouveau mécanisme d’inhibition qui pourrait être exploité comme alternative à la stratégie d’inhibition compétitive [3]. Ce mécanisme est illustré par la bréfeldine A (BFA), une toxine naturelle qui inhibe l’activation d’Arf, une protéine G impliquée dans le couplage entre le trafic et la morphologie cellulaires (pour revue, voir [4]). La BFA n’empêche pas l’interaction d’Arf avec son GEF, mais au contraire les piège dans une interaction non fonctionnelle [5]. Le mécanisme d’action de la BFA exploite les caractéristiques biochimiques de l’échange nucléotidique catalysé par les GEF qui sont communes à l’ensemble des protéines G et leurs GEF. La protéine G associée au GDP est d’abord reconnue dans un complexe de faible affinité par son GEF, puis des changements de conformation de grande amplitude conduisent à un complexe dépourvu de nucléotide et d’affinité élevée, auquel se fixe finalement le GTP qui dissocie le complexe (Figure 1, centre). La spécificité de l’interaction protéine G/GEF se joue avant la dissociation du GDP, car, dans le contexte cellulaire où le GTP est plus abondant que le GDP, la réaction d’échange ne peut plus faire marche arrière une fois que le GDP est dissocié. De façon remarquable, c’est à ce complexe ternaire de faible affinité que se fixe la BFA, piégeant un complexe Arf-GDP-BFA-GEF abortif incapable d’évoluer vers le complexe sans nucléotide (Figure 1, droite).
Nous avons étudié le mécanisme d’action de la BFA par cristallographie aux rayons X dans un complexe de protéines humaines comprenant la protéine G Arf1 et le domaine catalytique (appelé domaine Sec7) du facteur d’échange ARNO, ce dernier portant des mutations qui le rendent hypersensible à la toxine [3]. La structure à haute résolution du complexe montre que la BFA s’intercale à l’interface entre la protéine G et son facteur d’échange ; elle interagit pour 70 % avec la protéine G, et pour 30 % avec le facteur d’échange (Figure 2, gauche). Le GDP est toujours fixé dans le site nucléotidique où il conserve les interactions qui déterminent son affinité élevée, mais surtout il se trouve hors de portée du glutamate catalytique du GEF qui dissocie le nucléotide par répulsion électrostatique. La BFA bloque les réarrangements structuraux de grande amplitude au cours desquels la protéine G « roule » sur le GEF, et empêche ainsi qu’une interaction productive entre le glutamate catalytique et le nucléotide puisse se produire.
Figure 2
Les étapes de l’activation de la protéine G Arf en 3 dimensions.

La position du glutamate catalytique du GEF ou de sa mutation en lysine est en orange. Les régions d’Arf qui changent de conformation au cours de son cycle GDP/GTP sont indiquées en couleur. Les flèches indiquent les rotations successives d’Arf par rapport au domaine Sec7, la réaction évoluant de sa forme la plus ouverte vers la forme fermée sans nucléotide. Encart : gros plan sur l’interaction de la BFA à l’interface entre Arf-GDP et le domaine Sec7. La structure du même domaine en l’absence d’Arf (bleu foncé) montre que la BFA ne déforme pas le GEF (adapté de [3] avec la permission de l’éditeur).
De façon tout à fait remarquable, le site de fixation de la BFA ne préexiste ni dans la protéine G ni dans le GEF isolés, mais se forme transitoirement au cours du mécanisme d’échange pour disparaître à l’état sans nucléotide. La BFA épouse cette cavité avec une remarquable complémentarité de forme et de nature chimique (Figure 2, encart). Ses interactions sont en grande partie hydrophobes, avec quelques liaisons hydrogène dont la nature électrostatique oriente l’inhibiteur dans la cavité. Ces interactions déterminent d’ailleurs la spécificité d’action de la BFA : la toxine cible préférentiellement les facteurs d’échange et les protéines Arf dont la séquence leur permet de former ces liaisons hydrogène. Dans les cellules humaines, les complexes Arf/GEF sensibles à la BFA interviennent dans la dynamique de l’appareil de Golgi mais pas dans le couplage trafic/morphologie à la membrane plasmique (pour revue, voir [6]).
Ce mécanisme d’inhibition non compétitive trouve une variante dans l’inhibition de l’activation de Arf par une permutation de charge du glutamate catalytique de son GEF (Figure 1, droite) [7]. Dans la même étude, nous avons résolu la structure cristalline d’un complexe Arf-GDP-GEF[Glu/Lys], dans lequel l’étape bloquée par la BFA est franchie, mais les interactions électrostatiques répulsives qui délogent le GDP sont remplacées par des interactions qui le stabilisent (Figure 2, centre). Dans cette seconde structure, le GDP est toujours fixé à Arf, mais se trouve maintenant à proximité du site catalytique. Par comparaison au complexe Arf/GEF sans nucléotide [8] (Figure 2, droite), l’interface entre Arf et le domaine Sec7 est plus ouverte, avec la mutation en lysine s’intercalant en quelque sorte comme un obstacle à sa fermeture.
Les facteurs d’échange nucléotidique : caractérisation comme cibles et stratégie d’inhibition interfaciale
L’activation des protéines G par leur GEF apparaît de plus en plus comme une étape privilégiée où interrompre spécifiquement la (sur)activation de protéines G dans toute une variété de contextes pathologiques. Outre l’oncogène p21Ras (pour revue, voir [9]), les protéines G de la famille Rho sont ainsi clairement impliquées dans à peu près toutes les étapes de la progression des tumeurs (pour revue, voir [10]). Néanmoins, le défi est d’identifier les connexions cellulaires qui sont cruciales pour les processus tumoraux, et, parmi ces derniers, ceux qui bénéficieraient d’une réduction d’activité des protéines Rho. Le cas de l’oncogène Tiam est à cet égard significatif : l’invalidation chez la souris du gène codant pour ce GEF, utilisé pour la protéine G Rac, induit une forte résistance aux tumeurs engendrées par des agents mutagènes, mais une agressivité plus élevée des tumeurs [11]. Une augmentation de l’activité des protéines Rho est également impliquée dans le développement de maladies cardiovasculaires majeures comme l’hypertension artérielle, l’athérosclérose ou l’hypertrophie cardiaque ([11] et pour revues, voir [12, 13]). L’identité des GEF impliqués reste cependant à découvrir. Récemment, des GEF bactériens pour les protéines Rho et Arf ont aussi été identifiés chez des bactéries intracellulaires, permettant aux pathogènes de détourner des machineries cellulaires à leur profit ([14] et pour revue, voir [15]).
Comment nos travaux, de nature fondamentale, peuvent-ils se décliner dans le contexte de la recherche de nouvelles molécules thérapeutiques ? Les structures de complexes abortifs protéine G/GEF que nous avons identifiées permettent d’intercepter les états intermédiaires d’une réaction cellulaire fondamentale avec une précision sans précédent, mais elles débouchent aussi sur un concept pratiquement inexploré pour l’inhibition de protéines G d’intérêt thérapeutique, que nous avons appelé « inhibition interfaciale » : une molécule pharmacologique se fixe à l’interface entre une protéine G et son GEF et empêche le complexe d’entreprendre les réarrangements structuraux qui aboutissent à la dissociation du GDP. Par sa double interaction avec la protéine G et son GEF, une telle molécule peut en principe être conçue ou sélectionnée pour être spécifique de ses deux partenaires, et donc cibler la protéine G et son GEF uniquement dans la voie cellulaire où ils sont simultanément en action [16].
Nos analyses structurales révèlent que les complexes intermédiaires de la réaction d’échange présentent des interfaces protéine-protéine incomplètes, qui achèvent de s’organiser à l’étape sans nucléotide grâce à l’extraordinaire plasticité structurale de la protéine G. Ces intermédiaires de conformation plus ouverte exposent donc un talon d’Achille susceptible d’être ciblé par des inhibiteurs interfaciaux. Dans le détail, la BFA et la permutation de charge piègent des complexes différents, qui se déclinent de façon distincte en termes de mécanismes d’inhibition. La BFA exploite une spécificité structurale de la sous-famille Arf et Arf-like des protéines G, par lesquelles ces protéines établissent une communication structurale entre leur site nucléotidique et la face opposée de la protéine orientée vers la membrane [17]. Elle peut donc servir de plate-forme pour la recherche d’analogues ou de mimétiques inhibant cette famille de protéines. Un candidat d’intérêt est Arf6, une protéine impliquée dans les mécanismes de motilité cellulaire associés au processus métastatique, et qui n’est pas ciblée par la BFA [6]. Au contraire, la permutation de charge intercepte un intermédiaire « ouvert » de la réaction d’échange qui est vraisemblablement général à tous les mécanismes d’activation des protéines G par leurs GEF. Un intermédiaire « ouvert » avait d’ailleurs été anticipé par modélisation pour les protéines G de la famille Rho/Rac et leurs GEF à domaine Dbl-homology [18]. Ces données suggèrent qu’il devrait être possible de concevoir ou de cribler des molécules se fixant à l’interface protéine G/GEF au voisinage du site nucléotidique, capables d’agir comme un obstacle à la fermeture et stabilisant ainsi le complexe intermédiaire dans une conformation abortive. Comme pour la BFA, une telle molécule pourrait être en contact avec les deux partenaires de la réaction, et agir avec une double spécificité ; le mécanisme de la BFA établit de plus que le complexe intermédiaire peut être une espèce moléculaire transitoire.
La découverte des mécanismes structuraux du fonctionnement des GEF fournit aujourd’hui une base rationnelle pour la conception de molécules inhibitrices. Nous avons intégré cet objectif au sein d’un regroupement de laboratoires académiques (GDR 2823 du CNRS) dont le projet est de valider les GEF des Rho et Arf comme cibles thérapeutiques (http://www.lebs.cnrs-gif.fr/cherfils/gdr.html). Grâce à la réunion de compétences complémentaires incluant synthèse organique, modélisation, biologie structurale, biochimie, biologie moléculaire, biologie cellulaire, protéomique et physiopathologie, le GDR va mettre en oeuvre des stratégies interdisciplinaires pour développer des inhibiteurs de GEF permettant d’interrompre sélectivement les voies de signalisation hyperactives dans des contextes pathologiques humains.
Appendices
Remerciements
Ces travaux ont été soutenus par le programme ACI « Cibles thérapeutiques (CNRS/Inserm) », par l’Association pour la Recherche contre le Cancer et le programme Human frontiers. Nous remercions L. Renault et B. Guibert (LEBS) pour les études structurales, et S. Pasqualato pour la préparation de la Figure 1.
Références
- 1. Chardin P, Antonny B, Cherfils, J. Activation des petites protéines G par leurs facteurs d’échange. Med Sci(Paris) 2000 ; 16 : 228-34.
- 2. Gregg D, Rauscher FM, Goldschmidt-Clermont PJ. Rac regulates cardiovascular superoxide through diverse molecular interactions : more than a binary GTP switch. Am J Physiol Cell Physiol 2003 ; 285 : C723-34.
- 3. Renault L, Guibert B, Cherfils J. Structural snapshots of the mechanism and inhibition of a guanine nucleotide exchange factor. Nature 2003 ; 426 : 525-30 .
- 4. Donaldson JG, Jackson CL. Regulators and effectors of the ARF GTPases. Curr Opin Cell Biol 2000 ; 12 : 475-82.
- 5. Peyroche A, Antonny B, Robineau S, et al. Brefeldin A acts to stabilize an abortive ARF-GDP-Sec7 domain protein complex : involvement of specific residues of the Sec7 domain. Mol Cell 1999 ; 3 : 275-85.
- 6. Donaldson JG. Multiple roles for Arf6: sorting, structuring, and signaling at the plasma membrane. J Biol Chem 2003 ; 278 : 41573-6.
- 7. Beraud-Dufour S, Robinneau S, Chardin P, et al. A glutamic finger in the guanine nucleotide exchange factor ARNO displaces Mg2+ and the beta-phosphate to destabilize GDP on ARF1. EMBO J 1998 ; 17 : 3651-9.
- 8. Goldberg J. Structural basis for activation of ARF GTPase : mechanisms of guanine nucleotide exchange and GTP-myristoyl switching. Cell 1998 ; 95 : 237-48.
- 9. Quilliam LA, Rebhun JF, Castro AF. A growing family of guanine nucleotide exchange factors is responsible for activation of Ras-family GTPases. Prog Nucleic Acids Res Mol Biol 2002 ; 71 : 391-444.
- 10. Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat Rev Cancer 2002 ; 2 : 133-42.
- 11. Malliri A, van der Kammen RA, Clark K, et al. Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature 2002 ; 417 : 867-71.
- 12. Sauzeau V, Rolli-Derkinderen M, Lehoux S, Loirand G, Pacaud P. Sildenafil prevents change in RhoA expression induced by chronic hypoxia in rat pulmonary artery. Circ Res 2003 ; 93 : 630-7.
- 13. Clerk A, Sugden PH. Small guanine nucleotide-binding proteins and myocardial hypertrophy. Circ Res 2000 ; 86 : 1019-23.
- 14. Nagai H, Kagan JC, Zhu X, et al. A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science 2002 ; 295 : 679-82.
- 15. Burridge K, Wennerberg K. Rho and Rac take center stage. Cell 2004 ; 116 : 167-80.
- 16. Chardin P, McCormick F. Brefeldin A : the advantage of being uncompetitive. Cell 1999 ; 97 : 153-5.
- 17. Pasqualato S, Renault L, Cherfils J. Arf, Arl, Arp and Sar proteins : a family of GTP-binding proteins with a structural device for « front-back » communication. EMBO Rep 2002 ; 3 : 1035-41.
- 18. Cherfils J. Structural mimicry of DH domains by Arfaptin suggests a model for the recognition of Rac-GDP by its guanine nucleotide exchange factors. FEBS Lett 2001 ; 507 : 280-4.
List of figures
Figure 1
Le cycle GDP/GTP des protéines G.
À gauche : les protéines G sont activées par l’échange du GDP par du GTP, qui est stimulé par les GEF à la réception de signaux d’activation en amont. Elles transmettent alors un message d’action en interagissant avec les partenaires spécifiques de leur conformation liée au GTP, appelés collectivement « effecteurs ». Le cycle est terminé par les « GAP », qui stimulent l’hydrolyse du GTP en GDP. Dans certains cas comme celui des protéines Arf et Rho, le cycle GDP/GTP est couplé à une translocation de la protéine G du cytosol à la membrane. Au milieu : la réaction biochimique de la dissociation du GDP stimulée par les GEF. Les complexes ternaires protéine G/nucléotide/GEF ont une affinité beaucoup plus faible que le complexe sans nucléotide. À droite : l’activation de la protéine G Arf par ses GEF à domaine Sec7 peut être inhibée par stabilisation de complexes abortifs Arf-GDP-GEF en présence de la toxine bréfeldine A ou d’une mutation ponctuelle du GEF.
Figure 2
Les étapes de l’activation de la protéine G Arf en 3 dimensions.
La position du glutamate catalytique du GEF ou de sa mutation en lysine est en orange. Les régions d’Arf qui changent de conformation au cours de son cycle GDP/GTP sont indiquées en couleur. Les flèches indiquent les rotations successives d’Arf par rapport au domaine Sec7, la réaction évoluant de sa forme la plus ouverte vers la forme fermée sans nucléotide. Encart : gros plan sur l’interaction de la BFA à l’interface entre Arf-GDP et le domaine Sec7. La structure du même domaine en l’absence d’Arf (bleu foncé) montre que la BFA ne déforme pas le GEF (adapté de [3] avec la permission de l’éditeur).