Abstracts
Résumé
Durant les dix dernières années, des progrès substantiels ont été réalisés dans la compréhension, en termes moléculaires, des surdités héréditaires congénitales ou d’apparition précoce. Cet article porte essentiellement sur les surdités isolées (i.e. non syndromiques), pour lesquelles le nombre de gènes responsables identifiés (36 à ce jour) s’accroît rapidement. En revanche, des difficultés inhérentes à l’analyse de liaison génétique, auxquelles s’ajoute l’implication de facteurs environnementaux, ont jusqu’à présent empêché la caractérisation des principaux gènes responsables de, ou prédisposant à, l’apparition d’une surdité tardive.
Summary
This article outlines recent advances in explaining hereditary deafness in molecular terms, focusing on isolated (i.e. nonsyndromic) hearing loss. The number of genes identified (36 to date) is growing rapidly. However, difficulties inherent in genetic linkage analysis, coupled with the possible involvement of environmental causes, have so far prevented the characterization of the main genes causative or predisposing to the late-onset forms of deafness.
Article body
Les surdités peuvent être classées d’après le degré de la perte auditive (légère, modérée, sévère, profonde) et d’après l’emplacement du défaut primaire. On distingue ainsi les surdités de transmission, liées à un défaut de l’oreille externe ou de l’oreille moyenne, et les surdités de perception ou neurosensorielles, liées à un défaut en un endroit quelconque entre l’oreille interne et le cortex cérébral auditif. Les surdités de transmission sont légères ou moyennes. Elles sont, dans la très grande majorité des cas, secondaires à des otites. Rarement, il peut s’agir de malformations congénitales de l’oreille (aplasie notamment), parfois intégrées dans un syndrome héréditaire. Certaines surdités de transmission, d’installation tardive, sont dues à une maladie de l’os labyrinthique, l’otospongiose, qui entraîne une fixation de l’étrier à la fenêtre ovale (voir ci-dessous). Bon nombre de surdités de transmission sont curables chirurgicalement. Les surdités de perception ont une forte prévalence (voir Encadré). La plupart résultent d’un défaut de l’oreille interne. Le degré de la perte auditive y est variable. La prise en charge d’un enfant sourd - sévère ou profond - est éducative, orthophonique et prothétique (par prothèse conventionnelle ou par implant cochléaire).
La cochlée, organe de l’audition des mammifères
L’oreille interne des mammifères (Figure 1A) comprend la cochlée, organe de l’audition, et cinq organes vestibulaires, impliqués dans l’équilibration. La cochlée humaine détecte les fréquences sonores entre 20Hz et 20kHz («Promenade autour de la cochlée», www.iurc.montp.inserm.fr/cric51/audition/start.htm). Les organes vestibulaires, quant à eux, réagissent aux accélérations linéaire ou angulaire.
Figure 1
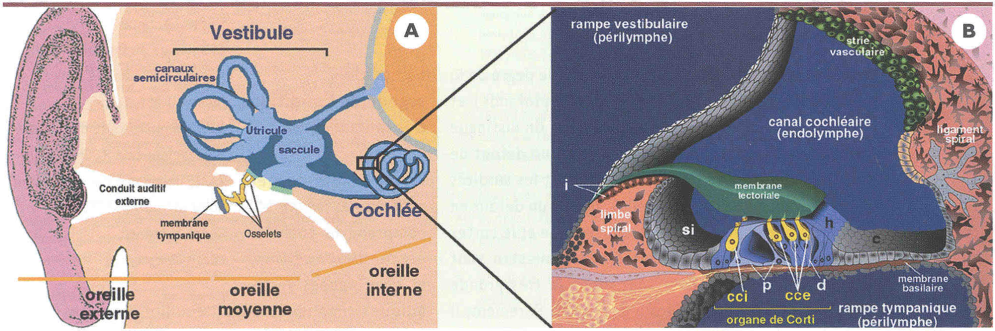
A. Représentation schématique de l’oreille humaine. L’oreille externe est constituée du pavillon et du conduit auditif externe, obturé par la membrane tympanique. L’oreille moyenne est une cavité aérienne qui comporte une chaîne de trois osselets (marteau, enclume, étrier). L’oreille interne comprend six organes sensoriels, la cochlée et cinq organes vestibulaires, le saccule, l’utricule et les trois canaux semi-circulaires. à la jonction entre l’utricule et le saccule se trouve le canal endolymphatique qui se termine par le sac endolymphatique. Cette excroissance intracrânienne du labyrinthe membraneux joue un rôle essentiel dans la résorption de l’endolymphe. B. Coupe transversale du canal cochléaire. Dans la cochlée, le labyrinthe membraneux délimite trois compartiments : les rampes vestibulaire et tympanique, toutes deux remplies de périlymphe, et le canal cochléaire, rempli d’endolymphe. L’organe de Corti, qui est l’appareil de la transduction mécano-électrique pour l’audition, fait saillie dans le canal cochléaire. Cet organe repose sur la membrane basilaire et est recouvert d’un gel acellulaire, la membrane tectoriale. Il est constitué des cellules sensorielles (en jaune), réparties en une rangée de cellules ciliées internes (cci) et trois rangées de cellules ciliées externes (cce), et de différents types de cellules de soutien, notamment les cellules piliers (p), les cellules de Deiters (d) et les cellules de Hensen (h). L’organe de Corti est flanqué par l’épithélium du sulcus interne (si), du côté médian, et par les cellules de Claudius (c), du côté latéral. La strie vasculaire, au niveau de la paroi latérale du canal cochléaire, est responsable de la sécrétion de K+ dans l’endolymphe et de la genèse du potentiel endocochléaire. Différents types de fibrocytes entourent l’épithélium bordant le canal cochléaire. i: cellules interdentales (adaptation d’une figure de P. Küssel-Andermann).
Une grande diversité de cellules épithéliales borde le labyrinthe membraneux de la cochlée (Figure 1B) [1]. L’épithélium sensoriel de la cochlée est appelé organe de Corti. Il repose sur la membrane basilaire et est recouvert d’un gel acellulaire, la membrane tectoriale. Il comprend deux types de cellules sensorielles ou cellules ciliées: une rangée unique de cellules ciliées internes, d’où part l’essentiel de l’influx nerveux en direction des centres auditifs, et une triple rangée de cellules ciliées externes. À l’apex de ces cellules, un ensemble de 30 à 300microvillosités rigides, appelées à tort stéréocils, constitue la structure de transduction mécano-électrique de la cellule ciliée [2]. Les stéréocils sont disposés en plusieurs rangées formant un escalier. L’extrémité de chaque stéréocil est reliée au stéréocil plus grand de la rangée voisine par un lien apical (Figure2). De plus, les stéréocils sont solidarisés en une touffe ciliaire par de nombreux liens latéraux. Des jonctions serrées assurent l’adhérence étroite de la partie apicale des cellules ciliées aux cellules de soutien voisines, formant une barrière imperméable aux liquides et solutés. Ainsi, alors que la touffe ciliaire baigne dans l’endolymphe, liquide riche en potassium (~150 mM) et pauvre en sodium (~1 mM) et en calcium (~0,02 mM), le reste de la cellule ciliée baigne dans la périlymphe, liquide pauvre en potassium (~3,5 mM) et riche en sodium (~140 mM) [3]. La strie vasculaire est un épithélium sécrétoire de la paroi latérale du canal cochléaire. Elle est constituée de deux couches principales de cellules, que sépare un espace liquidien contenant une couche discontinue de cellules intermédiaires et un réseau dense de capillaires sanguins. Les cellules basales de la strie vasculaire sont reliées aux cellules intermédiaires et aux fibrocytes du tissu conjonctif sous-jacent (ligament spiral) par des jonctions communicantes (gap junctions), ce qui indique l’existence d’échanges ioniques et métaboliques entre ces trois types de cellules. La strie vasculaire sécrète des ions K+ dans l’endolymphe, et produit une différence de potentiel électrique transépithéliale d’environ +80 mV, appelée potentiel endocochléaire, entre les compartiments endolymphatique et périlymphatique de la cochlée.
Figure 2
Les stéréocils et leurs liens apicaux.
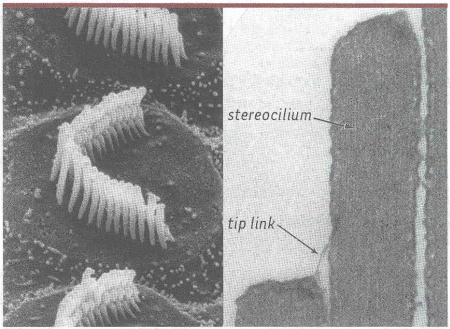
A. Photographie en microscopie électronique à balayage de la touffe ciliaire d’une cellule ciliée externe. B. Détail de deux stéréocils appartenant à des rangées adjacentes. Le lien apical (tip link), qui relie l’apex de chaque stéréocil à la paroi latérale du stéréocil plus grand de la rangée voisine, est visible.
L’onde aérienne de pression qui constitue le son est transmise aux liquides de l’oreille interne via la membrane tympanique, les trois osselets de l’oreille moyenne (marteau, enclume, étrier) et la membrane de la fenêtre ovale. La vibration de la membrane basilaire qui en résulte est maximale à un emplacement qui dépend de la fréquence [4]. Ainsi, il existe une organisation spatiale, ou tonotopie, de l’analyse des différentes fréquences constitutives d’un son: les fréquences les plus faibles (graves) sont analysées à l’apex de la cochlée, alors que les fréquences les plus élevées (aigus) sont analysées à la base de la cochlée. La vibration de la membrane basilaire en un endroit donné est transmise aux cellules ciliées sus-jacentes. Le mouvement relatif de la surface apicale de la cellule ciliée par rapport à la membrane tectoriale défléchit la touffe ciliaire. La tension des liens apicaux augmente, ce qui a pour effet d’ouvrir les canaux de la transduction mécano-électrique. Le flux entrant de potassium, sous l’effet du gradient de potentiel électrique d’environ 150mV entre le compartiment endolymphatique et le cytoplasme de la cellule ciliée, entraîne une dépolarisation membranaire de la cellule ciliée. Dans les cellules ciliées internes, qui sont les véritables cellules sensorielles, la dépolarisation entraîne une libération du neurotransmetteur (glutamate). Les fibres nerveuses afférentes transmettent alors un train de potentiels d’action codant pour les caractéristiques principales du stimulus sonore: intensité, durée et fréquence. L’organisation tonotopique s’étend aux voies nerveuses auditives jusqu’aux aires auditives du cortex cérébral. Les cellules ciliées externes jouent un rôle essentiel dans la sensibilité de l’organe auditif des mammifères [5]. Dans ces cellules, la dépolarisation entraîne une contraction rapide de la paroi latérale (transduction électro-mécanique). Ces changements cycliques de la longueur des cellules ciliées externes seraient responsables de l’amplification du stimulus acoustique par un effet de résonance avec la vibration de la membrane basilaire. L’analyse spectrale du son par l’oreille des mammifères repose donc à la fois sur les caractéristiques biophysiques de la membrane basilaire et sur la contractilité des cellules ciliées externes.
Causes des surdités syndromiques et non syndromiques
En dehors des embryopathies dues à la rubéole, la toxoplasmose ou l’infection par le cytomégalovirus, qui peuvent conduire à des polymalformations avec surdité, les surdités syndromiques sont d’origine génétique. On a décrit plusieurs centaines de syndromes qui associent à la surdité des anomalies très variées [6]. Les gènes responsables de plus de 100 syndromes ont été identifiés à ce jour (pour une liste de ces syndromes et des gènes correspondants, voir chapitre 254 de la version actualisée de MMBID, genetics.accessmedicine. com/) [7].
Les formes non syndromiques de surdité peuvent être causées par des facteurs environnementaux et/ou génétiques. Dans les pays développés, plus de deux tiers des cas de surdité isolée prélinguale ont une origine génétique. À de rares exceptions près, toutes les formes héréditaires de surdité analysées jusqu’à présent correspondent à des atteintes monogéniques (pour revue, voir [8]). Cependant, l’hétérogénéité phénotypique qui est le plus souvent observée pour une mutation donnée suggère fortement la contribution de gènes modificateurs [9-12]. On a longtemps pensé que les formes tardives de surdité résultaient d’une combinaison de causes génétiques et environnementales. Le nombre croissant de familles identifiées dans lesquelles la surdité se révèle tardivement, indique que la contribution des facteurs génétiques a été sous-estimée. En particulier, ces facteurs seraient importants pour l’otospongiose [13].
Génétique moléculaire des surdités non syndromiques
Les formes isolées de surdité héréditaire sont classées selon leur mode de transmission: DFN, DFNA et DFNB, désignent respectivement des formes de surdité transmises sur le mode lié au chromosome X, autosomique dominant et autosomique récessif. Pour chaque groupe, les locus sont numérotés dans l’ordre chronologique d’identification (par exemple, DFNB1, DFNB2...). Environ 80% des cas de surdité prélinguale correspondent à des formes autosomiques récessives, alors que la plupart des formes tardives correspondent à des formes dominantes.
L’identification des gènes responsables de surdité n’a véritablement commencé que dans les années 1990, en raison des obstacles que constituaient pour la cartographie chromosomique, l’extrême hétérogénéité génétique[2] de ce handicap, l’absence de signes cliniques distinctifs pour les différentes atteintes géniques à l’origine de surdité, et la fréquence des unions entre individus malentendants ou issus de malentendants. Ces difficultés ont pu être surmontées grâce au recrutement et à l’analyse de familles atteintes de surdité, vivant dans des isolats géographiques [8]. À ce jour, 71locus de surdité ont été identifiés (37 pour les formes DFNA, 34 pour les formes DFNB, 4 pour les formes DFN, 2correspondant à des anomalies du génome mitochondrial). Pour 43 formes génétiques, les gènes responsables (36 gènes) ont été identifiés (Tableau I). Ils codent en particulier pour des protéines impliquées dans le contrôle de la transcription, la structure ou la dynamique du cytosquelette des cellules sensorielles, la contractilité des cellules ciliées externes, les jonctions intercellulaires, la structure de la membrane tectoriale ou encore les flux d’ions et de métabolites qui sont échangés entre deux cellules ou entre une cellule et le milieu extracellulaire [14]. Mais cette liste est loin d’être exhaustive et on trouve également des protéines impliquées dans d’autres processus cellulaires plus ou moins bien définis. Enfin, pour plusieurs gènes responsables de surdité, la fonction de la protéine est totalement inconnue.
Tableau I
Gènes de surdité isolée identifiés chez l’homme et les mutants murins correspondants.

1surdité syndromique ; 2mutant obtenu par invalidation génique; 3mutant obtenu par invalidation ciblée dans les épithéliums sensoriels cochléaire et vestibulaire.
La découverte, dans les cas de surdité prélinguale, de la forte prévalence des mutations bi-alléliques du gène de la connexine 26 dans les pays du pourtour méditerranéen [8] revêt un intérêt médical particulier. En France, environ un tiers des cas de surdité congénitale profonde sont dus à l’anomalie de ce gène. Le développement du diagnostic moléculaire correspondant a permis d’améliorer considérablement la qualité du conseil génétique pour les individus sourds et leur famille. Un autre progrès important au plan médical est la découverte de l’association de mutations particulières du gène codant pour l’ARN ribosomique mitochondrial 12S à la surdité induite par les antibiotiques du groupe des aminoglycosides [8], ce qui devrait permettre de réduire fortement l’incidence de cette forme de surdité iatrogène.
Il est à noter qu’il existe plusieurs exemples dans lesquels une forme récessive et une forme dominante de surdité résultent d’anomalies d’un même gène (Tableau I). Dans ces cas, les mutations impliquées dans la forme DFNA ont vraisemblablement un effet dominant négatif, puisque les porteurs hétérozygotes des mutations impliquées dans la forme DFNB correspondante n’ont généralement pas d’atteinte de l’audition.
Enfin, plusieurs gènes responsables de formes syndromiques de surdité sont également impliqués dans des formes de surdité isolée (Tableau I). Dans la plupart des cas, il n’existe pas de corrélation directe entre le type de mutation et l’existence d’une surdité syndromique ou non syndromique. En outre, les résultats d’une analyse phénotypique approfondie dans certaines familles affectées par le syndrome de Pendred (surdité et goitre thyroïdien) ou le syndrome de Usher (surdité et rétinopathie) [15], suggèrent qu’il pourrait exister une sorte de continuum phénotypique entre des formes syndromiques et non syndromiques de surdité. Dans ces cas, il est très probable que des gènes modificateurs contribuent à la variabilité du phénotype.
La souris au service de l’homme
De nombreuses lignées de souris mutantes atteintes d’un défaut isolé de l’oreille interne ont été décrites (www.ihr.mrc.ac.uk/hereditary/MutantsTable.shtml, www.jax.org/research/hhim) [16, 17]. Dans presque tous les cas, la surdité s’accompagne de signes de dysfonctionnement vestibulaire (mouvements de rotation, instabilité de la tête, incapacité à nager). Chez l’homme cependant, la surdité héréditaire n’est que rarement associée à des troubles de l’équilibre, probablement à cause d’une compensation cérébrale des anomalies vestibulaires périphériques, qui repose principalement sur les systèmes visuel et proprioceptif. Parmi les gènes murins responsables de surdité qui ont été identifiés, il en est certains pour lesquels le gène orthologue humain n’a pas encore été impliqué dans une surdité (Tableau II). Dans les cas où l’orthologue humain est également responsable de surdité, ces lignées de souris mutantes représentent des modèles précieux pour étudier les processus pathogéniques qui interviennent dans les formes correspondantes de surdité humaine (Tableau I). Un autre intérêt des modèles murins de surdité est la possibilité d’identifier des gènes modificateurs du déficit auditif grâce aux croisements de lignées de souris sourdes avec des lignées de fonds génétique différent, générant, parmi les descendants, des animaux porteurs d’un déficit auditif amoindri ou au contraire aggravé, et chez lesquels le degré de la surdité est transmis comme un caractère héréditaire [18-20].
Tableau II
Gènes responsables de surdité isolée chez la souris, non encore impliqués dans une surdité humaine.

* Mutant obtenu par invalidation génique.
Perspectives
Le cadre de la recherche génétique et clinique sur les surdités héréditaires est désormais en place. Le prochain défi est de développer des stratégies thérapeutiques alternatives aux prothèses auditives utilisées actuellement. Les tentatives futures pour empêcher la progression de la surdité, ou même la faire régresser, se fonderont vraisemblablement sur des approches diverses (thérapie médicamenteuse, cellulaire ou génique), mais quel que soit le procédé finalement retenu pour une forme de surdité donnée, l’étude des processus pathogéniques en cause apparaît comme un pré-requis. On peut fonder des espoirs sur la recherche de gènes modificateurs à effet antagoniste des mutations du «gène de surdité». Enfin, il apparaît urgent de développer des systèmes pour administrer des médicaments et pour transférer des gènes ou des cellules spécifiquement dans l’oreille interne. Ces recherches sont susceptibles d’avoir également des répercussions sur la thérapie des formes non héréditaires de surdité.
Appendices
Notes
-
[1]
La gamme d’intensité sonore que l’oreille humaine peut traiter correspond à un rapport de puissance de 10 millions pour 1. Pour cette raison, il est plus commode de décrire l’intensité du son en utilisant une échelle logarithmique et en exprimant un son sous la forme d’un rapport de deux intensités, l’une étant choisie comme la référence. L’unité de mesure utilisée est le Bel, qui correspond à un rapport d’intensité de 10:1. De plus, il est apparu souhaitable de comparer la sensibilité auditive d’un individu donné à des valeurs «normales». Dans ce but, le seuil d’audition est exprimé par rapport à une valeur de référence déterminée à partir d’un groupe de sujets jeunes ayant une audition normale. Lorsque l’audition est testée avec un audiomètre en clinique, le seuil mesuré est exprimé en dB HL (decibels hearing level).
-
[2]
Une maladie monogénique est dite génétiquement hétérogène lorsqu’elle peut être causée par des mutations de gènes différents.
Références
- 1. Dallos P, Popper AN, Fay RR. The cochlea. New York: Springer, 1996.
- 2. Dulon D, Aran JM. Aspects cellulaires et moléculaires de la transduction mécano-sensorielle dans l’oreille interne. Med Sci (Paris) 1990 ; 6 : 744-54.
- 3. Sterkers O, Ferrary E, Tran Ba Huy P. Production des liquides de l’oreille interne. Med Sci (Paris) 1990 ; 6 : 755-61.
- 4. Von Békésy G. Experiments in hearing. New York: McGraw-Hill, 1960.
- 5. Dallos P, Fakler B. Prestin, a new type of motor protein. Nat Rev Mol Cell Biol 2002 ; 3 : 104-11.
- 6. Gorlin RJ. Hereditary hearing loss and its syndromes. New York: Oxford University Press, 1995.
- 7. Petit C, Levilliers J, Marlin S, Hardelin JP. Hereditary hearing loss. In : Scriver CR, Beaudet AL, Sly WS, Valle D, eds The metabolic and molecular bases of inherited disease. New York : McGraw-Hill, 2001 : 6281-328.
- 8. Petit C, Levilliers J, Hardelin JP. Molecular genetics of hearing loss. Annu Rev Genet 2001 ; 35 : 589-646.
- 9. Friedman T, Battey J, Kachar B, et al. Modifier genes of hereditary hearing loss. Curr Opin Neurobiol 2000 ; 10 : 487-93.
- 10. Riazuddin S, Castelein CM, Ahmed ZM, et al. Dominant modifier DFNM1 suppresses recessive deafness DFNB26. Nat Genet 2000 ; 26 : 431-4.
- 11. Nadeau JH. Modifier genes in mice and humans. Nat Rev Genet 2001 ; 2 : 165-74.
- 12. Haider NB, Ikeda A, Naggert JK, Nishina PM. Genetic modifiers of vision and hearing. Hum Mol Genet 2002 ; 11 : 1195-206.
- 13. Chen W, Campbell CA, Green GE, et al. Linkage of otosclerosis to a third locus (OTSC3) on human chromosome 6p21.3-22.3. J Med Genet 2002 ; 39 : 473-7.
- 14. Gilgenkrantz S. K+, le Seigneur des anneaux... de la cochlée. Med Sci (Paris) 2000 ; 16 : 270-2.
- 15. Petit C. Usher syndrome: from genetics to pathogenesis. Annu Rev Genom Hum Genet 2001 ; 2 : 271-97.
- 16. Steel KP. Inherited hearing defects in mice. Annu Rev Genet 1995 ; 29 : 675-701.
- 17. Anagnostopoulos AV. A compendium of mouse knockouts with inner ear defects. Trends Genet 2002 ; 18 : S21-38.
- 18. Johnson KR, Zheng Qing Y, Bykhovskaya Y, et al. A nuclear-mitochondrial DNA interaction affecting hearing impairment in mice. Nat Genet 2001 ; 27 : 191-4.
- 19. Ikeda A, Zheng QY, Zuberi AR, et al. Microtubule-associated protein 1A is a modifier of tubby hearing (moth1). Nat Genet 2002 ; 30 : 401-5.
- 20. Noben-Trauth K, Zheng QY, Johnson KR. Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nat Genet 2003 ; 35 : 21-3.
List of figures
Figure 1
A. Représentation schématique de l’oreille humaine. L’oreille externe est constituée du pavillon et du conduit auditif externe, obturé par la membrane tympanique. L’oreille moyenne est une cavité aérienne qui comporte une chaîne de trois osselets (marteau, enclume, étrier). L’oreille interne comprend six organes sensoriels, la cochlée et cinq organes vestibulaires, le saccule, l’utricule et les trois canaux semi-circulaires. à la jonction entre l’utricule et le saccule se trouve le canal endolymphatique qui se termine par le sac endolymphatique. Cette excroissance intracrânienne du labyrinthe membraneux joue un rôle essentiel dans la résorption de l’endolymphe. B. Coupe transversale du canal cochléaire. Dans la cochlée, le labyrinthe membraneux délimite trois compartiments : les rampes vestibulaire et tympanique, toutes deux remplies de périlymphe, et le canal cochléaire, rempli d’endolymphe. L’organe de Corti, qui est l’appareil de la transduction mécano-électrique pour l’audition, fait saillie dans le canal cochléaire. Cet organe repose sur la membrane basilaire et est recouvert d’un gel acellulaire, la membrane tectoriale. Il est constitué des cellules sensorielles (en jaune), réparties en une rangée de cellules ciliées internes (cci) et trois rangées de cellules ciliées externes (cce), et de différents types de cellules de soutien, notamment les cellules piliers (p), les cellules de Deiters (d) et les cellules de Hensen (h). L’organe de Corti est flanqué par l’épithélium du sulcus interne (si), du côté médian, et par les cellules de Claudius (c), du côté latéral. La strie vasculaire, au niveau de la paroi latérale du canal cochléaire, est responsable de la sécrétion de K+ dans l’endolymphe et de la genèse du potentiel endocochléaire. Différents types de fibrocytes entourent l’épithélium bordant le canal cochléaire. i: cellules interdentales (adaptation d’une figure de P. Küssel-Andermann).
Figure 2
Les stéréocils et leurs liens apicaux.
A. Photographie en microscopie électronique à balayage de la touffe ciliaire d’une cellule ciliée externe. B. Détail de deux stéréocils appartenant à des rangées adjacentes. Le lien apical (tip link), qui relie l’apex de chaque stéréocil à la paroi latérale du stéréocil plus grand de la rangée voisine, est visible.
List of tables
Tableau I
Gènes de surdité isolée identifiés chez l’homme et les mutants murins correspondants.
1surdité syndromique ; 2mutant obtenu par invalidation génique; 3mutant obtenu par invalidation ciblée dans les épithéliums sensoriels cochléaire et vestibulaire.
Tableau II
Gènes responsables de surdité isolée chez la souris, non encore impliqués dans une surdité humaine.
* Mutant obtenu par invalidation génique.