Abstracts
Résumé
Le profil hémodynamique du choc septique, influencé par de multiples variations physiologiques induites par l’infection, est caractérisé par l’association à des degrés variables de composantes hypovolémique, obstructive, cardiogénique, distributive ou cytotoxique. Ce profil est modifié par la restauration volémique, mais la persistance d’une hypotension signe le choc septique. Même si le débit cardiaque semble normal, voire élevé, des arguments solides témoignent d’une altération des propriétés contractiles du myocarde. Si le dysfonctionnement purement myocardique est rarement la cause directe du décès, il peut largement y contribuer. Faute d’une connaissance suffisante des phénomènes qui concourent à son développement, l’utilisation de substances visant à le corriger peut parfois s’avérer délétère. Une intégration plus précise de la physiopathologie du dysfonctionnement myocardique au cours du choc septique devrait permettre d’améliorer sa prise en charge.
Summary
Myocardial dysfunction frequently accompanies severe sepsis and septic shock. It is now clear that such a myocardial depression, as evidenced by biventricular alteration, is present during the early phase of sepsis in most patients. Myocardial depression exists despite a fluid loading-dependent hyperdynamic state and usually recovers within 7 to 10 days in survivors. Myocardial dysfunction does not appear to be due to irreversible structural abnormalities nor to myocardial hypoperfusion, but rather linked to many circulating mediators including cytokines. At a cellular level, reduced myocardial contractility could be related in part to apoptosis and induced by both nitric oxide-dependent and nitric oxide-independent mechanisms. However, whatever the mechanism involved, it leads to calcium homeostasis abnormality. The present review describes both the diagnosis procedure and the molecular and cellular pathways of sepsis-induced myocardial depression.
Article body
Le sepsis constitue un syndrome traduisant la réponse inflammatoire systémique à une infection. Lorsqu’il s’accompagne d’une défaillance cardiocirculatoire, il devient « choc septique » et la mortalité croît de 20 % à 40-70 %. Ces chiffres ont peu varié au cours des dernières années, malgré d’importantes avancées dans les domaines de l’antibiothérapie et des techniques de réanimation [1].
Le tableau hémodynamique observé au cours du choc septique est typiquement caractérisé, après expansion volémique, par un état hyperdynamique avec effondrement des résistances vasculaires périphériques, associé à un débit cardiaque normal ou élevé et à une pression artérielle abaissée. Néanmoins, il existe de nombreux arguments démontrant l’existence d’un dysfonctionnement myocardique intrinsèque dès la phase aiguë du sepsis.
Difficulté d’évaluation de la fonction cardiaque dans le sepsis
Quels indices pour évaluer la fonction systolique ventriculaire gauche ?
Les anomalies circulatoires constatées au cours du choc septique rendent difficile la mise en évidence d’un dysfonctionnement myocardique autonome, indépendant des conditions de charge : le débit cardiaque n’est pas un bon indice de contractilité ventriculaire, et sa normalité n’exclut en rien l’existence d’une altération des performances systoliques ventriculaires [2]. La fraction d’éjection du ventricule gauche (FEVG) n’est pas non plus un reflet parfait de la contractilité myocardique : elle dépend certes de la contractilité myocardique, mais aussi de la compliance myocardique, de la précharge et de la postcharge [3].
L’utilisation d’indices indépendants des conditions de charge a permis de démontrer l’existence d’une altération des propriétés contractiles ventriculaires au cours du sepsis [2, 4]. Sur les boucles pression-volume ventriculaire tracées pour différents niveaux de postcharge, les points télésystoliques forment une droite traduisant la relation pression-volume télésystolique du ventricule gauche. La pente de cette droite, appelée élastance maximale (Emax), est un bon indice de contractilité ventriculaire, indépendant des conditions de charge. Il a été démontré, dans les modèles animaux de choc septique [6], mais aussi chez l’homme, que la pente de cette relation pression-volume télésystolique est déplacée en bas et à droite (diminution de Emax), témoignant d’une contractilité diminuée. Cette méthode d’évaluation nécessite plusieurs adaptations pour pouvoir être utilisée en pratique clinique : le volume télésystolique ventriculaire gauche peut être apprécié par la surface télésystolique en échographie et la pression ventriculaire gauche par la pression aortique centrale ou fémorale (Figure 1). En utilisant cette technique, A. Cariou et al. [5] ont présenté l’évolution de l’élastance du ventricule gauche chez des patients en choc septique traités par dobutamine (Figure 2).
Figure 1
Reconstruction off-line des boucles pression-surface du ventricule gauche.

Deux signaux doivent être enregistrés simultanément (A) : la pression artérielle aortique, à l’aide d’un cathéter artériel inséré par voie fémorale, et la surface ventriculaire gauche, à l’aide d’un capteur transoesophagien (ETO) et d’un appareil d’échocardiographie doté d’un logiciel de détection automatique des contours (DAC). Après acquisition et correction du décalage temporel d’acquisition, les deux signaux obtenus (B) sont interfacés et vont permettre la reconstruction off-line de boucles pression-surface nécessaires au calcul de l’élastance télésystolique.
Figure 2
Exemple de boucles pression-surface obtenues chez un patient.
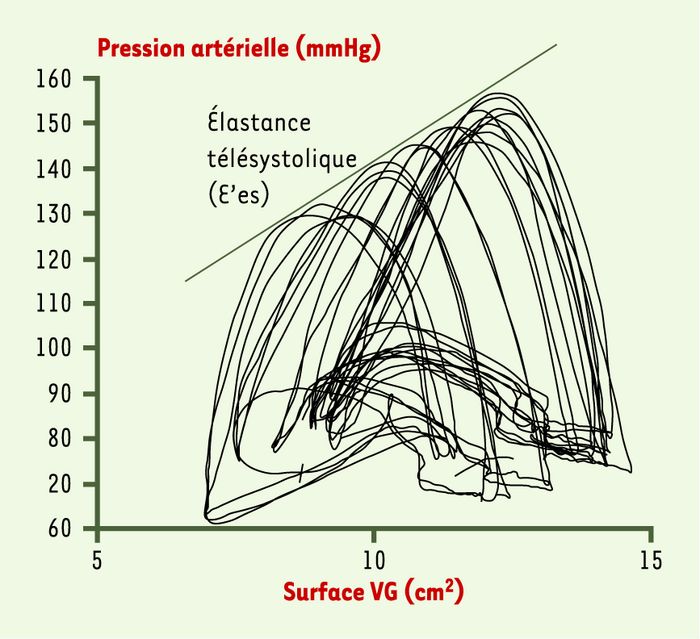
Lors d’une manoeuvre thérapeutique susceptible de faire varier rapidement, mais très transitoirement, le niveau de pré- et/ou de postcharge, le déplacement des points télésystoliques des boucles pression-surface s’effectue le long d’une droite dont la pente caractérise à un temps donné la valeur de l’élastance télésystolique du ventricule étudié (end-systolic elastance ou E’es).
La survenue d’une dilatation ventriculaire gauche au cours du choc septique reste source de controverse. Cette dilatation expliquerait pour certains le caractère « adapté » de la réponse en débit observée au cours du choc septique : elle correspondrait alors à un mécanisme compensateur susceptible d’offrir aux ventricules la capacité de maintenir un débit cardiaque adapté grâce à la loi de Franck-Starling (Figure 3) [2, 6]. La réalité de cette dilatation ventriculaire est toutefois incomplètement démontrée [6].
Figure 3
Évolution de la contractilité myocardique au cours du sepsis.

L’élastance ventriculaire gauche constitue l’indice de contractilité le plus indépendant des conditions de charge. L’exemple montré correspond à trois enregistrement successifs réalisés chez un même patient souffrant d’un choc septique en rapport avec une infection à pneumocoque. Comme on peut le voir, l’élastance E’es s’améliore très nettement entre la phase aiguë (J1) et la phase de récupération (J5, puis J8).
Le ventricule droit est-il également en cause ?
La plupart des études de la fonction ventriculaire droite au cours du sepsis ont été réalisées en utilisant la fraction d’éjection du ventricule droit (FEVD) comme indice de contractilité ventriculaire droite en thermodilution. Cette technique a permis de démontrer l’existence d’un dysfonctionnement ventriculaire droit corrélé à celui du ventricule gauche, et d’évolution parallèle [7].
Mécanismes du dysfonctionnement myocardique du sepsis
Anomalies structurelles
C.J. Fernandes et al. [8] ont mis en évidence plusieurs anomalies structurelles dans une étude autopsique de 71 patients décédés de choc septique. Par ailleurs, une augmentation fréquente des concentrations circulantes de troponine I (TnI) a été mise en évidence au cours du choc septique [9] : ces concentrations étaient corrélées à l’importance du dysfonctionnement myocardique et à la mortalité. L’oedème du tissu cardiaque lié à l’hyperperméabilité capillaire peut également altérer l’élastance ventriculaire [10].
Apoptose
L’apoptose, ou mort cellulaire programmée, joue un rôle important dans la régulation physiologique de la maturation et de la prolifération des cellules immunitaires, mais participe également du dysfonctionnement d’organe. Le TNF-α (tumor necrosis factor-α) ou l’endotoxine peuvent induire l’apoptose de cardiomyocytes de rats en culture [11]. L’inhibition pharmacologique de l’activation des caspases prévient l’apoptose et le dysfonctionnement cardiaques en cas de sepsis [11].
Facteurs humoraux
L’injection d’endotoxine bactérienne chez le volontaire sain reproduit toutes les caractéristiques hémodynamiques du choc septique, incluant une altération de la fonction systolique appréciée sur la FEVG [12]. Quant aux cytokines inflammatoires (TNF-α, interleukines 1, 2 et 6, MIF, macrophage migration inhibitory factor), leur injection chez l’animal et le volontaire sain peut reproduire ou contribuer à reproduire les effets cliniques du choc septique [13, 14].
Rôle du monoxyde d’azote (NO)
L’induction de l’enzyme NOS-2 (nitric oxide synthase) au cours du choc septique est responsable de la production importante de NO qui explique une partie des manifestations vasculaires périphériques (vasodilatation et hypotension). Sur le myocarde lui-même, les effets du NO sont cependant moins clairs : physiologiquement, le NO est produit en petites quantités au niveau du myocarde sous l’effet de NOS-3, présente de manière constitutive au niveau de l’endothélium coronarien. Le NO exercerait des effets paracrines facilitant la relaxation active et la compliance myocardique, mais aurait peu d’effets sur les caractéristiques contractiles [15]. La plupart des effets myocardiques du NO sont liés à l’activation de la guanylate-cyclase soluble, responsable de la production de GMP cyclique (GMPc) qui diminue les effets du calcium cytosolique sur la contraction filamentaire. Le GMPc est ainsi capable de stimuler certaines phosphodiestérases responsables de la dégradation de l’AMP cyclique (AMPc). La diminution du taux d’AMPc intracellulaire expliquerait la capacité du NO à atténuer les effets de la stimulation β-adrénergique. Pourtant, l’hypocontractilité de cardiomyocytes issus d’animaux endotoxinémiques n’est pas réversible ex vivo par l’administration d’inhibiteurs de la NOS [16].
Homéostasie calcique intracellulaire et couplage excitation-contraction
Dans des modèles d’animaux endotoxinémiques, une diminution du nombre de canaux calciques sarcolemmiques et de l’intensité du courant calcique lent a été notée [17]. L’évolution de cette diminution de courant calcique est corrélée à l’intensité du dysfonctionnement myocardique in vivo [17].
Concernant les altérations de la réponse myocardique à la stimulation α-adrénergique, les données sont très discordantes : des anomalies en termes de densité d’expression de surface, de fonctionnalité, ou des deux ont été rapportées [17, 18]. L’existence de ces anomalies renforce l’intérêt de l’utilisation d’agents inotropes n’empruntant pas la voie d’action des récepteurs β et notamment les agents capables de renforcer la sensibilité des myofilaments au Ca2+ tels que le lévosimendan [19].
Thérapeutique
Les objectifs cliniques usuels du traitement du sepsis peuvent, dans un premier temps, être atteints par l’administration d’un remplissage vasculaire adéquat. La correction de la vasodilatation excessive nécessite souvent l’utilisation d’amines vasopressives. Néanmoins, ces catécholamines pourraient induire une nécrose myocardique localisée avec inflammation périvasculaire. La vasopressine semble être une alternative séduisante [20, 21].
Conclusions
L’existence d’un dysfonctionnement myocardique transitoire au cours du sepsis est désormais un fait établi. Cette altération des performances myocardiques peut être mise en évidence à l’aide d’indices indépendants des conditions de charge. Les mécanismes concourant à sa survenue font intervenir des éléments de la réponse inflammatoire systémique nombreux et largement intriqués. Si le dysfonctionnement purement myocardique est rarement la cause directe du décès, il peut grandement y participer.
Appendices
Références
- 1. Friedman G, Silva E, Vincent JL. Has the mortality of septic shock changed with time? Crit Care Med 1998 ; 26 : 2078-86.
- 2. Parker MM, Shelhamer JH, Bacharach SL, et al. Profound but reversible myocardial depression in patients with septic shock. Ann Intern Med 1984 ; 100 : 483-9.
- 3. Robotham JL, Takata M, Berman M, Harasawa Y. Ejection fraction revisited. Anesthesiology 1991 ; 74 : 172-83.
- 4. Ognibene FP, Parker MM, Natanson C, et al. Depressed left ventricular performance. Response to volume infusion in patients with sepsis and septic shock. Chest 1988 ; 93 : 903-10.
- 5. Cariou A, Vinsonneau C, Chiche D, et al. L’élastance ventriculaire gauche est-elle altérée au cours du sepsis ? Réanim Urgences 2000 ; 9 (suppl 2) : 91S.
- 6. Poelaert J, Declerck C, Vogelaers D, et al. Left ventricular systolic and diastolic function in septic shock. Intensive Care Med 1997 ; 23 : 553-60.
- 7. Vincent JL, Reuse C, Frank N, et al. Right ventricular dysfunction in septic shock: Assessment by measurements of right ventricular ejection fraction using the thermodilution technique. Acta Anesthesiol Scand 1989 ; 33 : 34-8.
- 8. Fernandes CJ Jr, Iervolino M, Neves RA, et al. Interstitial myocarditis in sepsis. Am J Cardiol 1994 ; 74 : 958
- 9. Turner A, Tsamitros M, Bellomo R. Myocardial cell injury in septic shock. Crit Care Med 1999 ; 27 : 1775-80.
- 10. Yu P, Boughner DR, Sibbald WJ, et al. Myocardial collagen changes and edema in rats with hyperdynamic sepsis. Crit Care Med 1997 ; 25 : 657-62.
- 11. Neviere R, Fauvel H, Chopin C, et al. Caspase inhibition prevents cardiac dysfunction and heart apoptosis in a rat model of sepsis. Am J Respir Crit Care Med 2001 ; 163 : 218-25.
- 12. Taylor FB, Haddad PA, Hack E, et al. Two-stage response to endotoxin infusion into normal human subjects: correlation of blood phagocyte luminescence with clinical and laboratory markers of the inflammatory, hemostatic response. Crit Care Med 2001 ; 29 : 326-34.
- 13. Dinarello CA. Proinflammatory and anti-inflammatory cytokines as mediators in the pathogenesis of septic shock. Chest 1997 ; 112 (suppl 6) : S321-9.
- 14. Lesur O, F Chagnon, B Lévy, et al. Myocardial dysfunction in experimental septic shock: Effect of macrophage inhibitory factor (MIF) neutralization. Am J Respir Crit Care Med 2002 ; 165 : A176.
- 15. Paulus WJ, Vantrimpont PJ, Shah AM. Paracrine coronary endothelial control of left ventricular function in humans. Circulation 1995 ; 92 : 2119-26.
- 16. Tavernier B, Li JM, El-Omar MM, et al. Cardiac contractile impairment associated with increased phosphorylation of troponine I in endotoxemic rats. FASEB J 2001 ; 15 : 294-6.
- 17. Abi-Gerges N, Tavernier B, Mebazaa A, et al. Sequential changes in autonomic regulation of cardiac myocytes after in vivo endotoxin injection in rats. Am J Respir Crit Care Med 1999 ; 160 : 1196-204.
- 18. Tavernier B, Abi-Gerges N, Mebazaa A. Alteration of beta-adrenergic pathway in the septic heart. In : Vincent JL, ed. Yearbook of intensive care and emergency medicine. Berlin : Springer-Verlag, 1999 : 504-18.
- 19. Ming MJ, Hu DY, Chen HS, et al. Effects of MCI-154, a calcium sensitizer, on calcium sensitivity of myocardial fibers in endotoxic shock rats. Shock 2000 ; 14 : 652-6.
- 20. Landry DW, Levin HR, Gallant EM, et al. Vasopressin deficiency contributes to vasodilatation of septic shock. Circulation 1997 ; 95 : 1122-5.
- 21. Dunser MW, Mayr AJ, Ulmer H, et al. Arginine vasopressin in advanced vasodilatory shock. A prospective, randomized controlled study. Circulation 2003 ; 107 : 2313-9.
List of figures
Figure 1
Reconstruction off-line des boucles pression-surface du ventricule gauche.
Deux signaux doivent être enregistrés simultanément (A) : la pression artérielle aortique, à l’aide d’un cathéter artériel inséré par voie fémorale, et la surface ventriculaire gauche, à l’aide d’un capteur transoesophagien (ETO) et d’un appareil d’échocardiographie doté d’un logiciel de détection automatique des contours (DAC). Après acquisition et correction du décalage temporel d’acquisition, les deux signaux obtenus (B) sont interfacés et vont permettre la reconstruction off-line de boucles pression-surface nécessaires au calcul de l’élastance télésystolique.
Figure 2
Exemple de boucles pression-surface obtenues chez un patient.
Lors d’une manoeuvre thérapeutique susceptible de faire varier rapidement, mais très transitoirement, le niveau de pré- et/ou de postcharge, le déplacement des points télésystoliques des boucles pression-surface s’effectue le long d’une droite dont la pente caractérise à un temps donné la valeur de l’élastance télésystolique du ventricule étudié (end-systolic elastance ou E’es).
Figure 3
Évolution de la contractilité myocardique au cours du sepsis.
L’élastance ventriculaire gauche constitue l’indice de contractilité le plus indépendant des conditions de charge. L’exemple montré correspond à trois enregistrement successifs réalisés chez un même patient souffrant d’un choc septique en rapport avec une infection à pneumocoque. Comme on peut le voir, l’élastance E’es s’améliore très nettement entre la phase aiguë (J1) et la phase de récupération (J5, puis J8).