Abstracts
Résumé
La différenciation cellulaire repose sur l’acquisition d’une nouvelle identité cellulaire et donc d’un nouveau programme d’expression génique. Nous avons montré que Tpit, un facteur de transcription spécifique des cellules corticotropes et mélanotropes de l’hypophyse, est un régulateur positif de la différenciation de ces cellules, mais aussi un régulateur négatif de la lignée gonadotrope. Outre un mécanisme de choix mutuellement exclusifs lors de la différenciation, ces travaux suggèrent un modèle complet de différenciation hypophysaire à choix binaires. En accord avec l’expression exclusive de Tpit dans les cellules corticotropes, plusieurs mutations du gène humain TPIT ont été identifiées chez des patients atteints d’un déficit corticotrope isolé congénital.
Summary
Pituitary hormone-producing cells differentiate sequentially from a common epithelial primordium, Rathke’s pouch, under the combinatorial action of a subset of tissue- and cell-restricted transcription factors. Some factors have been implicated in early events of pituitary induction and morphogenesis while other factors like Pit-1 and SF-1 have been associated with differentiation of particular lineages. In POMC-expressing cells, Pitx1, NeuroD1 and Tpit were shown to be important for cell specific transcription of the POMC gene. Since Tpit is exclusively expressed in pituitary POMC-expressing lineages, the corticotrophs and melanotrophs, we investigated the TPIT gene coding sequences in 17 patients presenting with congenital isolated ACTH deficiency (IAD). We demonstrated that human TPIT gene mutations cause a neonatal onset form of IAD (8/11), but not juvenile forms of this deficiency (0/6). In the absence of glucocorticoid replacement, IAD can lead to neonatal death by acute adrenal insufficiency. To assess the importance of Tpit in pituitary differentiation and function, we produced Tpit-null mice. Concordant with the human phenotype, Tpit-null mice have IAD : plasma ACTH is greatly reduced in these mice, their plasma corticosterone is undetectable and the adrenals are hypoplastic. Analysis of the pituitary in Tpit-null mice revealed multiple roles of this factor in cell differentiation. First, Tpit is a positive regulator for POMC cell differentiation. Tpit is also a negative regulator of the pituitary gonadotroph fate. Thus, Tpit operates as a molecular switch to orient differentiation of a common precursor towards either POMC or gonadotroph fate. A binary choice model of pituitary cell differentiation is presented.
Article body
L’hypophyse coordonne les fonctions endocrines essentielles des vertébrés. Elle intègre les informations provenant du cerveau par l’intermédiaire de l’hypothalamus, contrôle le fonctionnement des glandes endocrines périphériques que sont la thyroïde, les surrénales et les gonades, et règle ainsi les processus vitaux du métabolisme, de la croissance, du comportement et de la reproduction. L’hypophyse est constituée de six lignées cellulaires définies par l’hormone qu’elles sécrètent : les cellules thyréotropes (TSH, thyroid stimulating hormone), somatotropes (GH, growth hormone), lactotropes (PRL, prolactine), gonadotropes (LH, luteinizing hormone ; FSH, follicle stimulating hormone), mélanotropes (αMSH, α-melanocyte stimulating hormone) et corticotropes (ACTH, adrenocorticotropic hormone). L’ACTH et l’αMSH sont deux protéines clivées à partir du même précurseur, la pro-opiomélanocortine (POMC) [1]. Chez la souris, l’hypophyse adulte est formée de trois lobes. La post-hypophyse est d’origine neuro-ectodermique. Les deux autres lobes dérivent de l’épithélium oral : ce sont le lobe intermédiaire, composé uniquement de cellules mélanotropes, et le lobe antérieur, contenant les autres lignées. Chez l’homme, le lobe intermédiaire disparaît durant le développement embryonnaire, et l’hypophyse ne contient pas de cellules mélanotropes [2].
Les cellules hypophysaires se différencient séquentiellement à partir d’un seul épithélium, la poche de Rathke [3]. Ce processus de différenciation est orchestré par des facteurs de transcription tels que Pitx1/2, Hesx1 (Rpx) et Lhx3/4 qui dirigent l’induction et la morphogenèse hypophysaire précoce, ainsi que par d’autres facteurs qui jouent un rôle dans la différenciation de lignées spécifiques [3]. Par exemple, le récepteur nucléaire SF1 (steroidogenic factor 1) est essentiel pour la différenciation des cellules gonadotropes et le facteur de transcription Pit1 est requis pour la différenciation des lignées thyréotropes, somatotropes et lactotropes [3, 4]. Dans les cellules corticotropes, Pitx1, NeuroD1/β2 et Tpit sont nécessaires pour l’histospécificité de la transcription du gène codant pour la POMC [5, 6]. Tpit, un facteur de transcription à boîte T, a initialement été identifié comme partenaire transcriptionnel du facteur à homéodomaine Pitx1. Chez la souris, son expression est restreinte aux deux lignées hypophysaires exprimant la POMC, les cellules corticotropes et les cellules mélanotropes [6].
Nous avons identifié plusieurs mutations du gène TPIT chez des patients atteints de déficit corticotrope isolé congénital, permettant de mieux définir cette entité clinique et d’y apporter une explication moléculaire [7]. Postulant que la souris serait un bon modèle du déficit corticotrope isolé congénital, nous avons produit une souris dont le gène Tpit a été invalidé. Outre le fait de présenter un phénotype très proche de celui observé chez l’homme, ce modèle murin a apporté des informations importantes concernant le développement et la différenciation hypophysaire [8].
Déficit corticotrope isolé congénital
Chez l’homme, la POMC possède trois sites d’expression correspondant à des fonctions biologiques distinctes : la POMC produite dans l’hypothalamus règle l’appétit, celle produite dans la peau contrôle la pigmentation et la POMC sécrétée par les cellules corticotropes hypophysaires est clivée en ACTH, hormone essentielle à la production des glucocorticoïdes par les corticosurrénales [9]. Des mutations dans le gène codant pour la POMC ont été identifiées chez des patients souffrant de symptômes en relation avec ces trois sites d’expression de la POMC : une obésité sévère, un défaut de pigmentation et un déficit en glucocorticoïdes [10]. Le déficit congénital isolé en ACTH ne peut donc pas être attribué à un défaut du gène codant pour la POMC. Cette entité clinique rare, diagnostiquée de la période périnatale à la puberté, peut être la cause de décès néonatal par insuffisance surrénalienne aiguë en l’absence de traitement. La physiopathologie et la description clinique du déficit corticotrope isolé congénital sont actuellement mal définies [11, 12]. Bien que le CRH (corticotropin releasing hormone) ou son récepteur aient été proposés comme candidats responsables de ce déficit [12], TPIT est le premier gène présentant une spécificité d’expression compatible avec le phénotype du déficit corticotrope isolé congénital. Ainsi, par l’analyse de la séquence codante du gène TPIT dans une série de 17 patients présentant un déficit corticotrope isolé congénital, nous avons montré que les mutations de TPIT sont associées à un déficit congénital isolé en ACTH dans 8 cas sur 11 pour lesquels ce déficit est précoce (néonatal). En revanche, lorsque ce déficit apparaît plus tardivement dans l’enfance, nous n’avons pas mis en évidence de mutations de TPIT (6/6 cas) [7]. Les patients sans mutation de TPIT n’avaient pas d’antécédent familial de déficit corticotrope (qu’il soit juvénile ou néonatal).
Les huit enfants présentant une mutation de TPIT sont issus de six familles non apparentées. Ils sont nés à terme de parents sains (avec une fonction corticotrope normale). Deux de ces enfants sont les cadets d’enfants mort-nés de cause inconnue. Le tableau clinique et les constantes biologiques de ces enfants sont donnés dans le Tableau I. Les symptômes d’insuffisance surrénalienne disparaissaient dans tous les cas avec la corticothérapie substitutive. L’autopsie du frère d’une de nos patientes (patient 7, Tableau I) décédé dans la période néonatale avait révélé une hypoplasie extrême des surrénales (« surrénales miniatures »), ainsi qu’une hypophyse morphologiquement normale mais dépourvue d’immunoréactivité à l’ACTH [13]. Aucun patient ne présentait d’anomalie de la pigmentation ou du comportement alimentaire, comme cela a été observé dans les mutations du gène codant pour la POMC. Il faut noter que les 11 patients atteints de déficit corticotrope isolé néonatal présentaient un phénotype similaire.
Tableau I
Présentation clinique des huit patients atteints de déficit corticotrope isolé congénital et porteurs d’une mutation du gène TPIT.
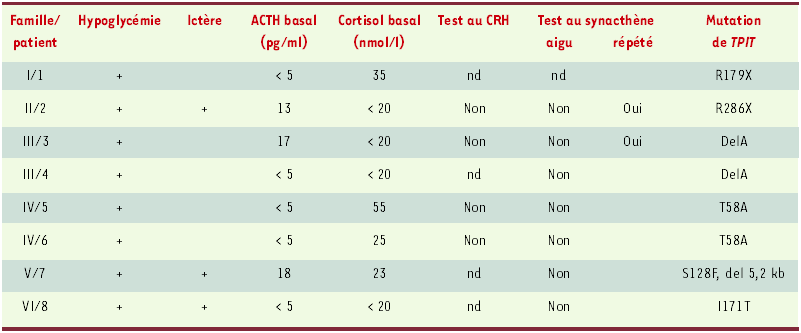
L’hypoglycémie néonatale était le plus souvent le premier symptôme ayant conduit au diagnostic de déficit en ACTH ; elle était le plus souvent sévère, quelquefois compliquée de convulsions. Trois patients ont présenté un ictère néonatal cholestatique prolongé. Tous les patients présentaient des concentrations plasmatiques effondrées d’ACTH (normale : 20-60 pg/ml) et de cortisol (normale : 250-600 nmol/l) ; les concentrations plasmatiques des autres hormones hypophysaires étaient normales dans tous les cas. La réponse au CRH de l’ACTH/cortisol (test au CRH) s’est révélée négative dans les quatre cas testés. L’injection aiguë de synacthène (test à l’ACTH) ne modifiait pas la concentration de cortisol plasmatique ; en revanche, les injections répétées de synacthène pendant 3 jours ont permis d’observer une réponse du cortisol dans deux cas, indiquant l’intégrité fonctionnelle des surrénales. nd : non déterminé.
Sept mutations ont été identifiées dans les exons codants de TPIT(Figure 1) : deux mutations non-sens (R179X, R286X), trois mutations faux-sens (T58A, I171T, S128F), une délétion d’une paire de base (nt782delA correspondant à la sérine 260 dans l’exon 6) et une large délétion de 5,2 kb comprenant les 3e et 4e exons du gène TPIT. Tous les patients étaient homozygotes pour leur mutation, sauf un patient qui était un hétérozygote composite (S128F et délétion des 3e et 4e exons). Tous les parents étaient hétérozygotes pour la mutation concernée, la majorité d’entre eux étant consanguins (5 familles sur 6). Ces observations sont en faveur d’un mode de transmission récessif. Les mutations non-sens (R179X, R286X) et la délétion d’une paire de base sont à l’origine de l’insertion d’un codon stop et entraîneraient une perte de fonction par dégradation de l’ARN messager [14]. Afin de déterminer la cause du déficit produit par les trois mutations faux-sens (T58A, I171T, S128F), nous avons inséré ces mutations dans des vecteurs d’expression et avons testé leur activité transcriptionnelle en transfections transitoires, ainsi que leur capacité de liaison à l’ADN. Les protéines mutantes ont une perte de leur activité transcriptionnelle, avec une absence de liaison à l’ADN [7]. Ces trois mutations affectent la boîte T de la protéine, notamment la thréonine en position 58 impliquée dans la liaison à l’ADN [15].
Figure 1
Mutations du gène TPIT dans les cas de déficit corticotrope isolé congénital.

Expression de TPIT dans les adénomes hypophysaires humains
L’expression de TPIT dans l’hypophyse humaine a été étudiée sur des fragments d’adénomes hypophysaires de différents types histologiques, recueillis par chirurgie trans-sphénoïdale [16]. L’étude immunohistochimique a montré que l’expression de TPIT était limitée au noyau des cellules corticotropes normales ou adénomateuses. Aucun immunomarquage n’a été observé dans les cellules d’adénomes lactotropes, somatotropes ou gonadotropes. En hybridation in situ, l’ARN messager de TPIT était co-exprimé avec celui de la POMC dans les cellules corticotropes normales et les adénomes corticotropes, mais n’était pas détectable dans les autres types d’adénomes hypophysaires. Ces résultats ont montré que TPIT était sélectivement exprimée dans les cellules corticotropes normales et adénomateuses chez l’homme, et représente donc un nouveau marqueur des cellules hypophysaires exprimant la POMC.
Souris dont le gène Tpit a été invalidé
Pour mieux comprendre le rôle de Tpit dans le développement des cellules exprimant la POMC, nous avons produit une souris dont le gène Tpit a été invalidé [8]. Comme les patients, les souris Tpit-/- présentent des concentrations plasmatiques effondrées d’ACTH et de glucocorticoïdes. En accord avec ces constatations biologiques, il existe une hypoplasie surrénalienne majeure prédominant sur la zone fasciculée. Contrairement aux patients porteurs de mutations de TPIT, la survie de ces souris homozygotes est similaire à celle des souris hétérozygotes et sauvages. De même, les souris Tpit-/- présentaient une glycémie normale à l’état basal. Toutefois, après 24 heures de jeûne, une hypoglycémie significativement plus marquée est observée chez les souris homozygotes. Une autre différence entre les phénotypes humain et murin est représentée par le défaut de pigmentation des souris Tpit-/-. La contribution en αMSH du lobe intermédiaire de la souris semble donc jouer un rôle important dans la pigmentation cutanée, tandis que chez l’homme qui, rappelons-le, n’a pas de lobe intermédiaire, la régulation de la pigmentation se fait uniquement au niveau de la peau.
L’analyse des hypophyses de souris adultes Tpit-/- a révélé un défaut de différenciation des cellules POMC, alors que le nombre des cellules des autres lignées est normal dans le lobe antérieur. Toutefois, ce défaut ne semble pas affecter le déclenchement de la différenciation corticotrope. En effet, durant le développement embryonnaire des souris Tpit-/-, un nombre normal de cellules exprimant NeuroD1 est observé. Ces cellules exprimant NeuroD1 représentent probablement des précurseurs corticotropes [5, 17]. Cependant, l’absence de Tpit empêche un développement complet de la lignée. Ces expériences suggèrent donc que Tpit est un régulateur positif pour la différenciation terminale des cellules corticotropes et mélanotropes et pour l’expression de POMC, mais n’est pas essentiel pour la détermination de ces lignées (Figure 2).
Figure 2
Changement de destinée cellulaire en l’absence de Tpit.

Comparée à celle des souris témoins (WT), l’hypophyse des souris Tpit-/- présente une forte diminution des cellules corticotropes et mélanotropes, avec une hypoplasie du lobe intermédiaire (LI). De plus, plusieurs cellules du lobe intermédiaire se différencient en cellules gonadotropes, ce qui semble indiquer un changement de destin cellulaire. LA : lobe antérieur ; LP : lobe postérieur.
Une analyse plus approfondie des hypophyses mutantes a permis de montrer que le lobe intermédiaire, qui ne contient normalement que des cellules mélanotropes, est non seulement hypoplasique, mais exprime de façon inattendue des marqueurs d’autres cellules hypophysaires. En effet, la sous-unité α des glycoprotéines (αGSU, α-glycoprotein subunit), commune à la TSH et aux gonadotrophines LH et FSH, et normalement restreinte aux cellules thyréotropes et gonadotropes, est exprimée dans plusieurs cellules du lobe intermédiaire des souris Tpit-/-. Parmi ces cellules, un très petit nombre exprime la βTSH, témoignant d’une différenciation thyréotrope, mais ces cellules n’expriment pas le facteur de transcription Pit1. En effet, durant le développement, deux populations de cellules thyréotropes sont engendrées : une lignée transitoire indépendante de Pit1 apparaît initialement dans la partie rostrale de la glande et disparaît à la naissance, et la population définitive de cellules thyréotropes dépendante de Pit1 apparaît secondairement dans la partie dorsale de la glande et demeure chez l’adulte [18].
Les marqueurs des cellules gonadotropes ont également été évalués. Les deux sous-unités des gonadotrophines LH et FSH (αGSU, βLH/βFSH), ainsi que le facteur SF1 - essentiel à leur différenciation - sont présents dans plusieurs cellules du lobe intermédiaire hypoplasique. Ces résultats suggèrent que Tpit pourrait être un régulateur négatif de la lignée gonadotrope (Figure 3). Nous avons confirmé cette hypothèse par des expériences de gain de fonction chez la souris dans lesquelles Tpit est exprimé sous le contrôle du promoteur de l’αGSU. Ainsi, la présence de Tpit dans les cellules gonadotropes bloque partiellement l’expression de SF1, de βFSH et d’αGSU et complètement celle de βLH. Nous avons aussi montré que Tpit bloque l’activité de SF1 agissant sur un élément de réponse à SF1, et vice versa. Ces résultats, de même que l’interaction in vitro de Tpit et SF1, sont en faveur d’un mécanisme de trans-répression pour rendre compte des activités opposées de Tpit et SF1.
Figure 3
Modèle binaire de différenciation des cellules hypophysaires chez la souris.

Dans ce modèle, les cellules corticotropes et mélanotropes (ACTH, αMSH) et les cellules gonadotropes (LH, FSH) proviennent d’un précurseur commun qui est différent du précurseur des lignées dépendantes du facteur de transcription Pit1 (GH, PRL, TSH). Dans les précurseurs cortico-, mélano- et gonadotropes, l’expression et l’antagonisme entre Tpit et SF1 établissent chacune des lignées respectives. Le facteur GATA-2 contribue à la différenciation des cellules gonadotropes.
Conclusions
Ces données suggèrent qu’un antagonisme entre Tpit et SF1, un facteur spécifique des cellules gonadotropes, pourrait jouer un rôle dans l’établissement des lignées POMC et gonadotropes et que ces lignées pourraient provenir de précurseurs communs (Figure 3). Ces travaux jettent un éclairage tout à fait nouveau sur les relations entre les lignées hypophysaires et permet de proposer un modèle de différenciation à choix binaire. Ce nouveau modèle propose des relations simples entre chacune des lignées hypophysaires. Le défi sera maintenant de mettre en évidence les précurseurs prédits par ce modèle.
Appendices
Remerciements
Nous remercions tous nos collègues cliniciens qui ont partagé leur matériel clinique avec nous : Michel David (Centre Hospitalier Lyon-Sud, Lyon, France), Georges Malpuech (Hôtel-Dieu, Clermont-Ferrand, France), Cheri Deal, Guy Van Vliet (Hôpital Sainte-Justine, Montréal, Canada), Monique De Vroede (University Medical Centre, Utrecht, Pays Bas), Felix G. Riepe, Carl-Joachim Partsch, Wolfgang G. Sippell (Division of Paediatric Endocrinology, Department of Paediatrics, Christian Albrechts University, Kiel, Allemagne), Merih Berberoglu, Begüm Atasay (Ankara University, Faculty of Medicine, Departments of Pediatric Endocrinology and Neonatology, Ankara, Turquie).
Références
- 1. Drouin J, Sun YL, Nemer M. Regulatory elements of the pro-opiomelanocortin gene. Pituitary specificity and glucocorticoid repression. Trends Endocrinol Metab 1990 ; 1 : 219-25.
- 2. Dubois PM, el Amraoui A, Heritier AG. Development and differentiation of pituitary cells. Microsc Res Tech 1997 ; 39 : 98-113.
- 3. Sheng HZ, Westphal H. Early steps in pituitary organogenesis. Trends Genet 1999 ; 15 : 236-40.
- 4. Parker KL, Rice DA, Lala DS, et al. Steroidogenic factor 1 : an essential mediator of endocrine development. Recent Prog Horm Res 2002 ; 57 : 19-36.
- 5. Poulin G, Lebel M, Chamberland M, et al. Specific protein : protein interaction between basic helix-loop-helix transcription factors and homeoproteins of the Pitx family. Mol Cell Biol 2000 ; 20 : 4826-37.
- 6. Lamolet B, Pulichino AM, Lamonerie T, et al. A pituitary cell-restricted T-box factor, Tpit, activates POMC transcription in cooperation with Pitx homeoproteins. Cell 2001 ; 104 : 849-59.
- 7. Pulichino AM, Vallette-Kasic S, Couture C, et al. Human and mouse Tpit gene mutations cause early onset pituitary ACTH deficiency. Genes Dev 2003 ; 17 : 711-6.
- 8. Pulichino AM, Vallette-Kasic S, Tsai JPY, et al. Tpit determines alternate fates during pituitary cell differentiation. Genes Dev 2003 ; 17 : 738-47.
- 9. Solomon S. POMC-derived peptides and their biological action. Ann NY Acad Sci 1999 ; 885 : 22-40.
- 10. Krude H, Biebermann H, Luck W, et al. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet 1998 ; 19 : 155-7.
- 11. Malpuech G, Vanlieferinghen P, Dechelotte P, et al. Isolated familial adrenocorticotropin deficiency : prenatal diagnosis by maternal plasma estriol assay. Am J Med Genet 1988 ; 29 : 125-30.
- 12. Kyllo JH, Collins MM, Vetter KL, et al. Linkage of congenital isolated adrenocorticotropic hormone deficiency to the corticotropin releasing hormone locus using simple sequence repeat polymorphisms. Am J Med Genet 1996 ; 62 : 262-7.
- 13. Dechelotte P, Darcha C, Labbe A, et al. Congenital adrenal hypoplasia due to isolated familial ACTH deficiency. Pediatr Pathol 1994 ; 14 : 377-80.
- 14. Hentze MW, Kulozik AE. A perfect message : RNA surveillance and nonsense-mediated decay. Cell 1999 ; 96 : 307-10.
- 15. Muller CW, Herrmann BG. Crystallographic structure of the T domain-DNA complex of the Brachyury transcription factor. Nature 1997 ; 389 : 884-8.
- 16. Vallette-Kasic S, Figarella-Branger D, Grino M, et al. Differential regulation of proopiomelanocortin and pituitary-restricted transcription factor (TPIT), a new marker of normal and adenomatous human corticotrophs. J Clin Endocrinol Metab 2003 ; 88 : 3050-6.
- 17. Poulin G, Turgeon B, Drouin J. NeuroD1/BETA2 contributes to cell-specific transcription of the POMC gene. Mol Cell Biol 1997 ; 17 : 6673-82.
- 18. Lin SC, Li S, Drolet DW, Rosenfeld MG. Pituitary ontogeny of the Snell dwarf mouse reveals Pit-1- independent and Pit-1-dependent origins of the thyrotrope. Development 1994 ; 120 : 515-22.
List of figures
Figure 1
Mutations du gène TPIT dans les cas de déficit corticotrope isolé congénital.
Figure 2
Changement de destinée cellulaire en l’absence de Tpit.
Comparée à celle des souris témoins (WT), l’hypophyse des souris Tpit-/- présente une forte diminution des cellules corticotropes et mélanotropes, avec une hypoplasie du lobe intermédiaire (LI). De plus, plusieurs cellules du lobe intermédiaire se différencient en cellules gonadotropes, ce qui semble indiquer un changement de destin cellulaire. LA : lobe antérieur ; LP : lobe postérieur.
Figure 3
Modèle binaire de différenciation des cellules hypophysaires chez la souris.
Dans ce modèle, les cellules corticotropes et mélanotropes (ACTH, αMSH) et les cellules gonadotropes (LH, FSH) proviennent d’un précurseur commun qui est différent du précurseur des lignées dépendantes du facteur de transcription Pit1 (GH, PRL, TSH). Dans les précurseurs cortico-, mélano- et gonadotropes, l’expression et l’antagonisme entre Tpit et SF1 établissent chacune des lignées respectives. Le facteur GATA-2 contribue à la différenciation des cellules gonadotropes.
List of tables
Tableau I
Présentation clinique des huit patients atteints de déficit corticotrope isolé congénital et porteurs d’une mutation du gène TPIT.
L’hypoglycémie néonatale était le plus souvent le premier symptôme ayant conduit au diagnostic de déficit en ACTH ; elle était le plus souvent sévère, quelquefois compliquée de convulsions. Trois patients ont présenté un ictère néonatal cholestatique prolongé. Tous les patients présentaient des concentrations plasmatiques effondrées d’ACTH (normale : 20-60 pg/ml) et de cortisol (normale : 250-600 nmol/l) ; les concentrations plasmatiques des autres hormones hypophysaires étaient normales dans tous les cas. La réponse au CRH de l’ACTH/cortisol (test au CRH) s’est révélée négative dans les quatre cas testés. L’injection aiguë de synacthène (test à l’ACTH) ne modifiait pas la concentration de cortisol plasmatique ; en revanche, les injections répétées de synacthène pendant 3 jours ont permis d’observer une réponse du cortisol dans deux cas, indiquant l’intégrité fonctionnelle des surrénales. nd : non déterminé.