Abstracts
Résumé
L’insuline joue un rôle anabolique majeur dans la mise en réserve des substrats glucidiques et lipidiques. Ses effets résultent de sa liaison à un récepteur membranaire spécifique exprimé en priorité sur ses trois tissus cibles, le foie, le muscle et le tissu adipeux. Ce récepteur possède une activité tyrosine-kinase qui permet une autophosphorylation du récepteur puis la phosphorylation sur des résidus tyrosine des protéines substats, protéines IRS (insulin receptor substrates) en priorité, et la création de complexes macromoléculaires d’activation à proximité du récepteur. Les deux voies majeures d’activation sont celles de la phosphatidylinositol-3 kinase, activant la protéine kinase B et impliquée en priorité dans les effets métaboliques, et la voie des MAP (mitogen-activated protein)-kinases, impliquée en priorité dans les effets nucléaires, la croissance et la différenciation. Cependant, l’activation d’un effet spécifique de l’insuline met fréquemment en jeu une conjonction de ces deux voies ainsi que d’autres voies intracellulaires, rendant ainsi compte de la pléiotropie et de la spécificité du signal. Le contrôle négatif du signal de l’insuline peut venir de la dégradation de l’hormone ou de la déphosphorylation du récepteur. Surtout, il va provenir de la phosphorylation de résidus sérine/thréonine sur le récepteur et les protéines IRS. Cette phosphorylation peut être activée par de nombreux acteurs impliqués en pathologie dans la résistance à l’insuline, comme un hyperinsulinisme, le TNFα (tumor necrosis factor α) ou les acides gras libres libérés en excès par le tissu adipeux et transformés dans la cellule en acylCoA (acyl coenzyme A). Un rôle délètère des molécules libérées par le tissu adipeux est proposé dans la genèse de l’insulinorésistance hépatique et musculaire présente dans le diabète de type 2, l’obésité et le syndrome métabolique.
Summary
Insulin has a major anabolic function leading to storage of lipidic and glucidic substrates. All its effects result from insulin binding to a specific membrane receptor which is expressed at a high level on the 3 insulin target tissues: liver, adipose tissue and muscles. The insulin receptor exhibits a tyrosine-kinase activity which leads, first, to receptor autophosphorylation and then to tyrosine phosphorylation of substrates proteins, IRS proteins in priority. This leads to the formation of macromolecular complexes close to the receptor. The two main transduction pathways are the phosphatidylinositol 3 kinase pathway activating protein kinase B which is involved in priority in metabolic effects, and the MAP kinase pathway involved in nuclear effects, proliferation and differentiation. However, in most cases, a specific effect of insulin requires the participation of the two pathways in a complex interplay which could explain the pleiotropy and the specificity of the insulin signal. The negative control of the insulin signal can result from hormone degradation or receptor dephosphorylation. However, the major negative control results from phosphorylation of serine/threonine residues on the receptor and/or IRS proteins. This phosphorylation is activated in response to different signals involved in insulin resistance, hyperinsulinism, TNFα or increased free fatty acids from adipose tissue, which are transformed inside the cell in acyl-CoA. A deleterious role for molecules issued from the adipose tissue is postulated in the resistance to insulin of the liver and muscles present in type 2 diabetes, obesity and metabolic syndrome.
Article body
L’insuline joue un rôle anabolique majeur au niveau de l’organisme, dans la mise en réserve et l’utilisation des substrats énergétiques, qu’ils soient glucidiques ou lipidiques: entrée de glucose, synthèse de glycogène et lipogenèse, inhibition de la glycogénolyse, de la néoglucogenèse et de la lipolyse. Elle exerce également des fonctions pléïotropes sur le métabolisme protéique (augmentation de la synthèse et inhibition de la protéolyse), la croissance, le contrôle de l’apoptose et le développement (Figure 1) [1]. L’ensemble de ces effets résulte de la liaison de l’hormone à un récepteur spécifique présent à la surface de toutes les cellules de l’organisme, mais exprimé surtout dans ses trois tissus cibles, le foie, le muscle et le tissu adipeux.
Figure 1
Effets pléïotropes de l’insuline.

En se fixant sur son récepteur spécifique, l’insuline exerce ses effets dans de nombreux tissus, ses trois principaux tissus cibles étant le foie, le tissu adipeux et les muscles.
Le récepteur de l’insuline (RI)
Il appartient à la famille des récepteurs de facteurs de croissance qui possèdent une activité tyrosine kinase dans leur domaine intracellulaire. Il constitue le chef de file de la famille des récepteurs formés de quatre sous-unités, dont l’autre membre important est le récepteur de l’IGF1 (insulin-like growth factor de type 1). Le RI est formé de deux chaînes α extracellulaires reliées par des ponts disulfure à deux chaînes β transmembranaires. On peut considérer le récepteur comme un hétérodimère préassocié dans la membrane. Chaque sous-unité α possède un domaine complet de liaison de l’hormone situé de part et d’autre d’une séquence riche en cystéines permettant l’établissement de ponts disulfure structurant ce domaine. Cependant, une seule molécule d’insuline, en se liant avec une haute affinité sur les deux sous-unités α, va permettre d’activer complètement le récepteur. Les sites vacants ne peuvent alors être occupés qu’avec une basse affinité par une autre molécule d’insuline, du fait de l’encombrement stérique du domaine de liaison par la première molécule, et ne participent pas à l’activation du récepteur.
Le récepteur IGF1 a une structure similaire à celle du récepteur de l’insuline et une homologie de l’ordre de 50%. La liaison de l’IGF1 sur le RI, ou l’inverse, se fait avec une affinité 100 à 1 000 fois plus faible. Cependant, les concentrations circulantes en IGF1, 100 fois supérieures à celles de l’insuline (l’IGF1 étant stocké dans le sérum sur des protéines de liaison), pourraient activer le RI [2]; de même, dans des situations pathologiques d’insulinorésistance extrême, des concentrations en insuline très élevées peuvent activer le récepteur IGF1. Enfin, alors que les hépatocytes et les adipocytes expriment une grande quantité de RI et peu de récepteurs IGF1, les myocytes expriment les deux, et la présence de récepteurs hybrides en quantité importante a été montrée dans ce tissu [3]: ces récepteurs auraient des caractéristiques de liaison assez proches de celles des récepteurs de l’IGF1.
Le RI est codé par un gène unique. Sa synthèse est sujette à un épissage alternatif qui aboutit à un récepteur possédant (forme longue, B) ou non (forme courte, A) une séquence correspondant à l’exon 11 et codant 12acides aminés à l’extrémité carboxy-terminale de la chaîne α. L’expression de ces deux isoformes est différente selon les tissus, le rôle physiologique de cette expression différentielle restant mal compris. De façon intéressante, l’isoforme A du RI, exprimée majoritairement pendant la vie foetale, serait capable de lier l’IGF2 avec une bonne affinité et représenterait le récepteur de ce facteur de croissance pour les tissus foetaux [4]. Dans les processus tumoraux, au cours desquels une sécrétion autocrine d’IGF2 est souvent observée, l’isoforme A serait également mise en jeu. Dans la dystrophie musculaire de Steinert, la résistance musculaire à l’insuline a pu être corrélée à une expression quasi exclusive de l’isoforme A, moins efficiente que l’isoforme B pour transmettre le signal de l’hormone [5].
Les deux sous-unités β ont chacune un domaine transmembranaire donnant au récepteur une mobilité latérale. En l’absence d’insuline, les sous-unités α exercent une contrainte inhibitrice et maintiennent le récepteur en configuration inactive. La liaison de l’hormone permet un rapprochement des deux sous-unités β et l’activation du récepteur. Chaque sous-unité β porte un domaine à activité tyrosine kinase intracellulaire possédant une boucle régulatrice qui occlut le site catalytique tyrosine kinase et le maintient à l’état inactif. Lors de l’activation du récepteur, la liaison de l’ATP sur son site consensus permet le dépliement de cette boucle et sa transphosphorylation (c’est-à-dire la phosphorylation d’une sous-unité β par l’autre) sur des résidus tyrosine (résidus 1146, 1150 et 1151). Le domaine tyrosine kinase est alors complètement activé et peut ainsi phosphoryler d’autres tyrosines présentes sur les chaînes β, conduisant à une autophosphorylation du récepteur, mais aussi sur des protéines substrats. La phosphorylation de la tyrosine 960 de la chaîne β, notamment, va jouer un rôle clé dans l’ancrage ultérieur des protéines substrats sur le récepteur; cette phosphorylation n’est toutefois pas toujours mise en évidence [2, 6].
Transmission du signal insulinique
La transmission du signal insulinique dans la cellule met en jeu des modules protéiques de reconnaissance présents sur les protéines substrats et capables de les positionner à proximité du récepteur activé. Au moins 9substrats intracellulaires communs aux récepteurs de l’insuline et de l’IGF1 ont été identifiés. La première famille, qui compte 4 membres, est celle des IRS (insulin receptor substrate); ses principaux représentants, IRS1 et IRS2, jouent des rôles complémentaires dans la signalisation de l’insuline [7]. L’une des principales voies de la signalisation insulinique est celle de la phosphatidyl-inositol 3 (PI3) kinase (Figure 2). L’effet de l’insuline sur le transport du glucose, qui représente sans doute l’un des effets les mieux étudiés de l’hormone, illustre la complexité du signal de l’insuline. Celle-ci est capable d’induire la translocation, d’un compartiment intracellulaire vers la membrane plasmique, de vésicules contenant les transporteurs GLUT4, présentes dans les cellules musculaires et les adipocytes [1, 2, 8]. Ce processus met en jeu, à côté de la voie PI3 kinase, plusieurs autres voies de signalisation (Figure3). Cette complexité permet de comprendre comment le signal insulinique peut être spécifique vis-à-vis du transport du glucose: en effet, différentes hormones ou facteurs activant la PI3 kinase, mais incapables d’activer les voies annexes, ne peuvent induire un tel effet sur le transport de glucose.
Figure 2
Principales voies de signalisation par l’insuline: voies PI3 kinase et MAP kinase.

Les protéines IRS (insulin receptor substrate) (en jaune) se positionnent au niveau de la face cytosolique de la membrane plasmique par leur domaine PH (domaine d’homologie avec la pleckstrine) qui reconnaît probablement des phospholipides membranai-res. Elles positionnent ainsi leur domaine PTB (phosphotyrosine binding), adjacent au domaine PH, en face de la tyrosine 960 du récepteur de l’insuline (RI) (en vert), et se fixent au RI sur la tyrosine 960 phosphorylée par l’intermédiaire de leur domaine PTB (Figure 2). IRS2 va en outre interagir avec le domaine tyrosine-kinase du RI. La moitié carboxy-terminale des protéines IRS se trouve alors à proximité du domaine tyrosine kinase du récepteur, qui phosphoryle des résidus tyrosines spécifiques sur les IRS. Les protéines IRS ainsi phosphorylées sont à leur tour reconnues par les domaines SH2 (src homology 2) de protéines relais (en violet), les principales étant la sous-unité régulatrice de la phosphatidyl-inositol 3 (PI3) kinase, les protéines adaptatrices Grb2 (growth factor receptor-bound protein 2) et CrkII, la tyrosine-kinase Fyn et la phosphotyrosine phosphatase SHP2 (SH2 domain protein tyrosine phosphatase-2). A. La PI3 kinase est l’une des protéines importantes activées par cette liaison des IRS1 et 2; elle phosphoryle en position 3 les phosphoinositides membranaires, créant ainsi des sites de reconnaissance pour d’autres kinases cellulaires telles que la protéine kinase B (PKB)/Akt ou la PDK1/2 (3-phosphoinositide-dependent protein kinase 1/2). La PKB activée par phosphorylation va à son tour phosphoryler et activer d’autres relais intracellulaires impliqués en priorité dans les effets métaboliques de l’hormone. La phosphorylation de la glycogène synthase 3 kinase (GSK3)-β favorise la synthèse de glycogène. Celle de la kinase p70rsk et du facteur 4E-BP1 (4E binding protein 1), via la kinase mTOR (mammalian target of rapamycin), participe à l’action de l’insuline sur la synthèse protéique en augmentant le niveau général de traduction. La voie PI3 kinase/PKB intervient également dans le contrôle négatif de l’expression génique: en phosphorylant les facteurs de transcription de la famille Forkhead, tels que FKHR, elle permet leur rétention dans le cytosol et les empêche d’activer, au niveau nucléaire, leurs gènes cibles tels que celui de l’enzyme clé de la néoglucogenèse, la phosphoénolpyruvate carboxykinase. Toujours par la voie PKB, l’insuline exerce un effet anti-apoptotique en phosphorylant et inhibant le facteur pro-apoptotique Bad [6, 7]. B. Au départ du récepteur de l’insuline, deux voies aboutissent à l’activation de la voie MAP kinase: via les protéines IRS, la liaison de l’adaptateur Grb2 sur des phosphotyrosines spécifiques permet d’activer le facteur d’échange nucléotidique SOS (son of sevenless) qui active la petite protéine G Ras dans la membrane plasmique en stimulant l’échange du GDP contre le GTP. Ras active la kinase Raf, qui phosphoryle alors et active la MAP kinase kinase (MEK) responsable de l’activation par phosphorylation des deux MAP kinases, ERK1 et 2 (extracellular signal-regulated kinase). Celles-ci vont activer la kinase p90rsk impliquée dans la synthèse protéique et vont entrer dans le noyau afin de phosphoryler et activer des facteurs de transcription tels que p62TCF impliqués dans la prolifération et la différentiation cellulaire. Une deuxième possibilité de mise en route de la voie MAP kinase (à gauche sur la figure) part du récepteur de l’insuline qui recrute sur la tyrosine 960 les protéines adaptatrices de la famille SHC (src homologous and collagen protein) (en jaune), elles-mêmes reconnues par la protéine Grb2 activant la voie Ras.
Il existe à côté de la voie PI3 kinase une autre voie importante de signalisation par l’insuline, la voie MAP kinase, qui est commune à de nombreux facteurs de croissance et permet, in fine, d’activer l’expression génique et la prolifération (Figure2). Les voies PI3 kinase/PKB et MAP kinase sont interconnectées entre elles et participent à l’activation l’une de l’autre.
Contrôle négatif du signal de l’insuline: dégradation de l’hormone et du récepteur, déphosphorylation des tyrosines
La fin du signal insuline implique la dégradation de l’hormone après internalisation des complexes insuline-récepteur dans les endosomes. La majorité des récepteurs est recyclée au niveau de la membrane, tandis que d’autres sont dégradés. Dans les conditions physiologiques, les récepteurs nouvellement synthétisés permettent de restaurer un nombre normal de récepteurs sur la cellule. En présence d’un hyperinsulinisme persistant, en revanche, les cycles d’internalisation/recyclage peuvent aboutir à une diminution du nombre de récepteurs à la surface, processus de down regulation participant de façon secondaire à l’installation d’un phénomène de résistance à l’insuline.
La déphosphorylation des résidus tyrosine du récepteur et des protéines IRS requiert des tyrosine phosphatases (PTPases): les phosphatases PTP1B, cytosoliques, et LAR (leukocyte common antigen-related molecule), membranaires, ont été impliquées dans ces processus, notamment PTP1B, présente sur les récepteurs intracellulaires en cours d’endocytose [2]. Une augmentation de l’activité PTPase dans les muscles des patients diabétiques a été observée, qui participerait à la résistance de ces tissus à l’insuline. Des inhibiteurs de ces enzymes apparaissent ainsi constituer des outils thérapeutiques prometteurs.
Résistance à l’insuline: rôle des phosphorylations en Ser/Thre
La phosphorylation des résidus sérine ou thréonine semble jouer, vis-à-vis du récepteur et des protéines IRS, un rôle antagoniste de celui de la phosphorylation des seuls résidus tyrosine, intervenant probablement de façon majeure dans les mécanismes de résistance à l’insuline. Cette phosphorylation des résidus sérine ou thréonine permettrait de mettre fin à l’activation physiologique du récepteur, son exacerbation en pathologie ayant en revanche un rôle déletère induisant une résistance à l’hormone.
Plusieurs études se sont récemment intéressées à la phosphorylation sur Ser/Thr des protéines IRS1 et 2, qui découplerait ces protéines du récepteur et arrêterait la transduction du signal insuline [6]. De nombreux signaux sont capables d’induire cette phosphorylation, tels les acides gras libres, le diacylglycérol, les acyl-CoA et le glucose, mais également des cytokines inflammatoires comme le TNF (tumor necrosis factor) α ou l’IL (interleukine) 1β et même l’insuline, tous agents responsables de résistance à l’insuline. Parmi les enzymes capables de phosphoryler les IRS en Ser/Thr, on trouve la PKC ζ, la kinase IKK β (inhibitor of nuclear factor κB kinase), la MAP kinase et surtout la Jun kinase (JNK), qui semble particulièrement intéressante. Ainsi, en phosphorylant la sérine 307 de l’IRS1 murin (sérine 312 chez l’homme), la JNK empêche l’interaction du domaine PTB de IRS1/2 avec la tyrosine 960 phosphorylée du RI et induit un état de résistance à l’insuline (Figure 4). Cette kinase est activée par l’insuline elle-même, ce qui pourrait expliquer l’insulinorésistance associée aux états d’hyperinsulinisme [10], mais aussi par le TNFα et les acides gras libres sécrétés par le tissu adipeux, qui sont impliqués dans la résistance à l’insuline en pathologie [7, 11, 12]. L’élévation des acides gras libres résultant d’une lipolyse accrue en cas de résistance à l’insuline de ce tissu pourrait conduire à une accumulation de diacylglycérol et d’acylCoA dans les muscles et le foie, conduisant à une activation de la PKC θ et à une phosphorylation de l’IRS1 sur Ser/Thr, inhibant ainsi le signal insuline, notamment pour le transport du glucose dans le muscle [13]. Les anomalies des taux d’adipocytokines, leptine et adiponectine, aggraveraient cette accumulation en inhibant l’oxydation intramitochondriale des acides gras dans ces tissus [14, 15].
Figure 3
Activation par l’insuline de l’entrée du glucose dans les cellules musculaires et les adipocytes.
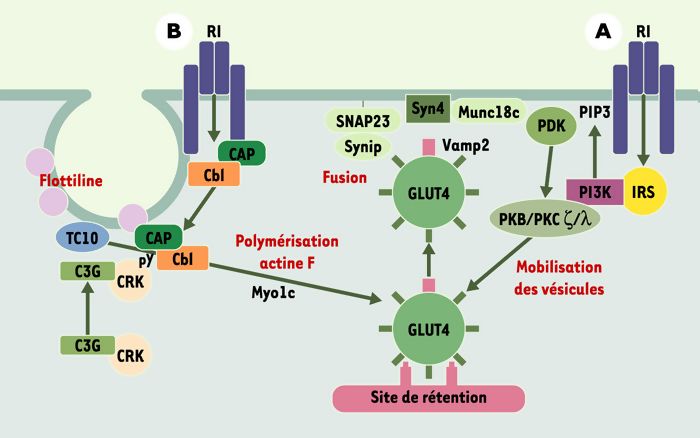
Une voie dépendante de la PI3 (phosphatidyl-inositol3) kinase (A), et passant par l’activation de la protéine kinase B (PKB) et de protéine kinases (PK) C atypiques, les PKC ζ/λ, va permettre de lever le signal de rétention qui maintenait les vésicules contenant les transporteurs du glucose GLUT4 au niveau du réseau transgolgien. Ces vésicules vont alors utiliser des microfilaments d’actine F, polymérisée en réponse à l’insuline via une voie mettant en jeu des intermédiaires spécifiques (B): la protéine CAP (c-Cbl associated protein), en se liant sur le récepteur de l’insuline activé, recrute la protéine c-Cbl phosphorylée sur un résidu tyrosine par le RI. Le complexe CAP-c-Cbl phosphorylé est ciblé vers des régions spécialisées de la membrane, les rafts (ou radeaux lipidiques), au niveau desquels il s’associe à la protéine flotilline (en rose). Cbl phosphorylée sur une tyrosine (pY) recrute alors l’adaptateur CrkII et le facteur d’échange nucléotidique C3G qui active la petite GTPase TC10, permettant la polymérisation de l’actine [8]. Le mouvement rapide des vésicules de GLUT4 le long des microfilaments d’actine met en jeu Myo1c, une protéine moteur moléculaire [9]. Enfin, il faut que les vésicules soient correctement ciblées vers la membrane, ce qui nécessite l’intervention de systèmes de reconnaissance vésicules/membrane cible de type v-SNARE/t-SNARE (vesicle or target SNAP (soluble NSF attachment protein) receptor): les vésicules de GLUT4 portent à leur surface des protéines VAMP 2 (vesicle associated membrane protein) 2 capables de reconnaître sur la membrane plasmique les protéines de la famille t-SNARE, syntaxine 4 et SNAP23, permettant ainsi la formation d’un complexe ternaire sous le contrôle de la protéine Munc 18c, puis la fusion avec la membrane et l’apport des transporteurs GLUT4. Cette reconnaissance nécessite, en réponse à l’insuline mais de façon indépendante de la voie PI3 kinase, la levée de l’inhibition exercée par la protéine Synip fixée sur Syn4, qui empêche la liaison du VAMP2 [1, 8].
Figure 4
Inhibition du signal insuline par phosphorylation sur Ser/Thr des protéines IRS.
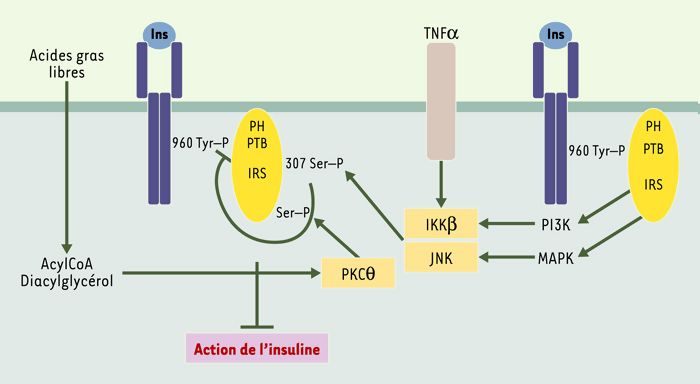
Cette phosphorylation peut résulter d’un rétrocontrôle du signal insuline, ou de l’action d’autres agents comme le TNFα (tumor necrosis factor α) et les acides gras libres, mais aussi de l’IL (interleukine) 1β, et même l’insuline. La kinase IKK β (inhibitor of nuclear factor κB kinase), la MAP-kinase et surtout la Jun kinase (JNK) sont capables d’effectuer de telles phosphorylations. En phosphorylant la sérine 307 de l’IRS1 murin, la JNK empêche l’interaction du domaine PTB de IRS1/2 avec la tyrosine 960 phosphorylée du RI et donc la transmission du signal insulinique. Cette kinase est activée par l’insuline et par le TNFα.Par ailleurs, l’élévation des acides gras libres et l’accumulation de diacylglycérol et d’acylCoA pourraient conduire à une activation de la PKC θ et à une phosphorylation de l’IRS1 sur Ser/Thr.
D’autres mécanismes sont également impliqués dans la régulation négative du signal insuline: la déphosphorylation des phosphoinositides par des lipide-phosphatases comme PTEN (phosphatase and tensin homolog deleted on chromosome ten) et SHIP (Src homology 2 domain-containing inositol 5 - phosphatase) réversent le signal PI3 kinase. Certaines protéines sont capables de se lier au récepteur et de bloquer la transmission du signal, comme les membres de la famille GRB7, 10 et 14 (cette dernière utilisant la protéine ZIP (protein kinase C ζ interacting protein) et la PKC ζ pour rétrocontrôler le RI) [16] et les protéines de la famille SOCS (suppressor of cytokine signaling), impliquées dans la régulation négative de la signalisation par les cytokines. La régulation de la quantité des protéines substrats IRS qui sont dégradées après ubiquitination va également contrôler la réponse à l’insuline.
Conclusions
Au total, le signal insuline emprunte dans la cellule des voies multiples interconnectées les unes aux autres, rendant compte de la pléïotropie et de la spécificité du signal qui va concerner non seulement le métabolisme énergétique, mais aussi la croissance et la différenciation cellulaires. Dans les situations pathologiques de résistance à l’insuline, le tissu adipeux joue un rôle important en raison de l’action des adipocytokines et des acides gras libres qu’il sécrète, qui vont bloquer la transmission du signal en différents points, notamment au niveau des protéines IRS jouant un rôle central dans l’activation, mais aussi dans l’inhibition des signaux hormonaux.
Appendices
Références
- 1. Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001; 414: 799-806.
- 2. Kido Y, Nakae J, Accili D. Clinical review 125: The insulin receptor and its cellular targets. J Clin Endocrinol Metab 2001; 86: 972-9.
- 3. Bailyes EM, Nave BT, Soos MA, Orr SR, Hayward AC, Siddle K. Insulin receptor/IGF-I receptor hybrids are widely distributed in mammalian tissues: quantification of individual receptor species by selective immunoprecipitation and immunoblotting. Biochem J 1997; 327: 209-15.
- 4. Frasca F, Pandini G, Scalia P, et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol 1999; 19: 3278-88.
- 5. Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet 2001; 29: 40-7.
- 6. Le Roith D, Zick Y. Recent advances in our understanding of insulin action and insulin resistance. Diabetes Care 2001; 24: 588-97.
- 7. White MF. IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab 2002; 283: E413-22.
- 8. Bryant NJ, Govers R, James DE. Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol 2002; 3: 267-77.
- 9. Bose A, Guilherme A, Robida SI, et al. Glucose transporter recycling in response to insulin is facilitated by myosin Myo1c. Nature 2002; 420: 821-4.
- 10. Lee YH, Giraud J, Davis RJ, White MF. cJUN N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J Biol Chem 2002; 278: 2896-902
- 11. Hirosumi J, Tuncman G, Chang L, et al. A central role for JNK in obesity and insulin resistance. Nature 2002; 420: 333-6.
- 12. Rui L, Aguirre V, Kim JK, et al. Insulin/IGF-1 and TNF-alpha stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J Clin Invest 2001; 107: 181-9.
- 13. Petersen KF, Shulman GI. Pathogenesis of skeletal muscle insulin resistance in type 2 diabetes mellitus. Am J Cardiol 2002; 90: G11-8.
- 14. Minokoshi Y, Kim YB, Peroni OD, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002; 415: 339-43.
- 15. Yamauchi T, Kamon J, Minokoshi Y, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 2002; 8: 1288-95.
- 16. Cariou B, Perdereau D, Cailliau K, et al. The adapter protein ZIP binds Grb14 and regulates its inhibitory action on insulin signaling by recruiting protein kinase Czeta. Mol Cell Biol 2002; 20: 6959-70.
List of figures
Figure 1
Effets pléïotropes de l’insuline.
En se fixant sur son récepteur spécifique, l’insuline exerce ses effets dans de nombreux tissus, ses trois principaux tissus cibles étant le foie, le tissu adipeux et les muscles.
Figure 2
Principales voies de signalisation par l’insuline: voies PI3 kinase et MAP kinase.
Les protéines IRS (insulin receptor substrate) (en jaune) se positionnent au niveau de la face cytosolique de la membrane plasmique par leur domaine PH (domaine d’homologie avec la pleckstrine) qui reconnaît probablement des phospholipides membranai-res. Elles positionnent ainsi leur domaine PTB (phosphotyrosine binding), adjacent au domaine PH, en face de la tyrosine 960 du récepteur de l’insuline (RI) (en vert), et se fixent au RI sur la tyrosine 960 phosphorylée par l’intermédiaire de leur domaine PTB (Figure 2). IRS2 va en outre interagir avec le domaine tyrosine-kinase du RI. La moitié carboxy-terminale des protéines IRS se trouve alors à proximité du domaine tyrosine kinase du récepteur, qui phosphoryle des résidus tyrosines spécifiques sur les IRS. Les protéines IRS ainsi phosphorylées sont à leur tour reconnues par les domaines SH2 (src homology 2) de protéines relais (en violet), les principales étant la sous-unité régulatrice de la phosphatidyl-inositol 3 (PI3) kinase, les protéines adaptatrices Grb2 (growth factor receptor-bound protein 2) et CrkII, la tyrosine-kinase Fyn et la phosphotyrosine phosphatase SHP2 (SH2 domain protein tyrosine phosphatase-2). A. La PI3 kinase est l’une des protéines importantes activées par cette liaison des IRS1 et 2; elle phosphoryle en position 3 les phosphoinositides membranaires, créant ainsi des sites de reconnaissance pour d’autres kinases cellulaires telles que la protéine kinase B (PKB)/Akt ou la PDK1/2 (3-phosphoinositide-dependent protein kinase 1/2). La PKB activée par phosphorylation va à son tour phosphoryler et activer d’autres relais intracellulaires impliqués en priorité dans les effets métaboliques de l’hormone. La phosphorylation de la glycogène synthase 3 kinase (GSK3)-β favorise la synthèse de glycogène. Celle de la kinase p70rsk et du facteur 4E-BP1 (4E binding protein 1), via la kinase mTOR (mammalian target of rapamycin), participe à l’action de l’insuline sur la synthèse protéique en augmentant le niveau général de traduction. La voie PI3 kinase/PKB intervient également dans le contrôle négatif de l’expression génique: en phosphorylant les facteurs de transcription de la famille Forkhead, tels que FKHR, elle permet leur rétention dans le cytosol et les empêche d’activer, au niveau nucléaire, leurs gènes cibles tels que celui de l’enzyme clé de la néoglucogenèse, la phosphoénolpyruvate carboxykinase. Toujours par la voie PKB, l’insuline exerce un effet anti-apoptotique en phosphorylant et inhibant le facteur pro-apoptotique Bad [6, 7]. B. Au départ du récepteur de l’insuline, deux voies aboutissent à l’activation de la voie MAP kinase: via les protéines IRS, la liaison de l’adaptateur Grb2 sur des phosphotyrosines spécifiques permet d’activer le facteur d’échange nucléotidique SOS (son of sevenless) qui active la petite protéine G Ras dans la membrane plasmique en stimulant l’échange du GDP contre le GTP. Ras active la kinase Raf, qui phosphoryle alors et active la MAP kinase kinase (MEK) responsable de l’activation par phosphorylation des deux MAP kinases, ERK1 et 2 (extracellular signal-regulated kinase). Celles-ci vont activer la kinase p90rsk impliquée dans la synthèse protéique et vont entrer dans le noyau afin de phosphoryler et activer des facteurs de transcription tels que p62TCF impliqués dans la prolifération et la différentiation cellulaire. Une deuxième possibilité de mise en route de la voie MAP kinase (à gauche sur la figure) part du récepteur de l’insuline qui recrute sur la tyrosine 960 les protéines adaptatrices de la famille SHC (src homologous and collagen protein) (en jaune), elles-mêmes reconnues par la protéine Grb2 activant la voie Ras.
Figure 3
Activation par l’insuline de l’entrée du glucose dans les cellules musculaires et les adipocytes.
Une voie dépendante de la PI3 (phosphatidyl-inositol3) kinase (A), et passant par l’activation de la protéine kinase B (PKB) et de protéine kinases (PK) C atypiques, les PKC ζ/λ, va permettre de lever le signal de rétention qui maintenait les vésicules contenant les transporteurs du glucose GLUT4 au niveau du réseau transgolgien. Ces vésicules vont alors utiliser des microfilaments d’actine F, polymérisée en réponse à l’insuline via une voie mettant en jeu des intermédiaires spécifiques (B): la protéine CAP (c-Cbl associated protein), en se liant sur le récepteur de l’insuline activé, recrute la protéine c-Cbl phosphorylée sur un résidu tyrosine par le RI. Le complexe CAP-c-Cbl phosphorylé est ciblé vers des régions spécialisées de la membrane, les rafts (ou radeaux lipidiques), au niveau desquels il s’associe à la protéine flotilline (en rose). Cbl phosphorylée sur une tyrosine (pY) recrute alors l’adaptateur CrkII et le facteur d’échange nucléotidique C3G qui active la petite GTPase TC10, permettant la polymérisation de l’actine [8]. Le mouvement rapide des vésicules de GLUT4 le long des microfilaments d’actine met en jeu Myo1c, une protéine moteur moléculaire [9]. Enfin, il faut que les vésicules soient correctement ciblées vers la membrane, ce qui nécessite l’intervention de systèmes de reconnaissance vésicules/membrane cible de type v-SNARE/t-SNARE (vesicle or target SNAP (soluble NSF attachment protein) receptor): les vésicules de GLUT4 portent à leur surface des protéines VAMP 2 (vesicle associated membrane protein) 2 capables de reconnaître sur la membrane plasmique les protéines de la famille t-SNARE, syntaxine 4 et SNAP23, permettant ainsi la formation d’un complexe ternaire sous le contrôle de la protéine Munc 18c, puis la fusion avec la membrane et l’apport des transporteurs GLUT4. Cette reconnaissance nécessite, en réponse à l’insuline mais de façon indépendante de la voie PI3 kinase, la levée de l’inhibition exercée par la protéine Synip fixée sur Syn4, qui empêche la liaison du VAMP2 [1, 8].
Figure 4
Inhibition du signal insuline par phosphorylation sur Ser/Thr des protéines IRS.
Cette phosphorylation peut résulter d’un rétrocontrôle du signal insuline, ou de l’action d’autres agents comme le TNFα (tumor necrosis factor α) et les acides gras libres, mais aussi de l’IL (interleukine) 1β, et même l’insuline. La kinase IKK β (inhibitor of nuclear factor κB kinase), la MAP-kinase et surtout la Jun kinase (JNK) sont capables d’effectuer de telles phosphorylations. En phosphorylant la sérine 307 de l’IRS1 murin, la JNK empêche l’interaction du domaine PTB de IRS1/2 avec la tyrosine 960 phosphorylée du RI et donc la transmission du signal insulinique. Cette kinase est activée par l’insuline et par le TNFα.Par ailleurs, l’élévation des acides gras libres et l’accumulation de diacylglycérol et d’acylCoA pourraient conduire à une activation de la PKC θ et à une phosphorylation de l’IRS1 sur Ser/Thr.