Abstracts
Résumé
Le diabète de type 2 - maladie de l’homéostasie du glucose - est un problème majeur de santé publique, qui se caractérise par deux anomalies majeures: une perturbation de la sécrétion des hormones pancréatiques (diminution quantitative et qualitative - phase précoce, pulsatilité - de la sécrétion d’insuline, augmentation de la sécrétion de glucagon), et une perturbation des effets de l’insuline sur ses tissus cibles (insulinorésistance). Les anomalies de l’action de l’insuline sur les tissus cibles se traduisent par une diminution du captage de glucose par les muscles et par une augmentation de la production hépatique de glucose. Elles sont liées à des défauts multiples dans les mécanismes de signalisation par le récepteur de l’insuline et dans des étapes régulatrices du métabolisme du glucose (transport, enzymes clés de la synthèse de glycogène ou de l’oxydation mitochondriale du glucose). Ces défauts «post-récepteurs» sont amplifiés par la présence d’une concentration augmentée d’acides gras libres. Les mécanismes impliqués dans les effets «diabétogènes» des acides gras libres sont analysés dans cet article. En effet, les concentrations élevées d’acides gras libres plasmatiques contribuent à la diminution de l’utilisation musculaire de glucose (principalement par l’atténuation de la transmission du signal insulinique) et à l’augmentation de la production hépatique (stimulation de la néoglucogenèse par l’apport de co-facteurs tels que l’acétyl-CoA, l’ATP et le NADH). L’exposition chronique à des concentrations élevées d’acides gras entraîne une accumulation d’acyl-CoA dans les cellules β du pancréas qui se traduit par la disparition de 50% de ces cellules par apoptose (phénomène de lipotoxicité).
Summary
Type 2 diabetes is characterized by two major defects: a dysregulation of pancreatic hormone secretion (quantitative and qualitative - early phase, pulsatility - decrease of insulin secretion, increase in glucagon secretion), and a decrease in insulin action on target tissues (insulin resistance). The defects in insulin action on target tissues are characterized by a decreased in muscle glucose uptake and by an increased hepatic glucose production. These abnomalities are linked to several defects in insulin signaling mechanisms and in several steps regulating glucose metabolism (transport, key enzymes of glycogen synthesis or of mitochondrial oxidation). These postreceptors defects are amplified by the presence of high circulating concentrations of free fatty acids. The mechanisms involved in the «diabetogenicity» of long-chain fatty acids are reviewed in this paper. Indeed, elevated plasma free fatty acids contribute to decrease muscle glucose uptake (mainly by reducing insulin signaling) and to increase hepatic glucose production (stimulation of gluconeogenesis by providing cofactors such as acetyl-CoA, ATP and NADH). Chronic exposure to high levels of plasma free fatty acids induces accumulation of long-chain acyl-CoA into pancreatic β-cells and to the death of 50 % of β-cell by apoptosis (lipotoxicity).
Article body
Les acides gras libres (AGL) sont les substrats énergétiques majeurs d’un très grand nombre de tissus (muscles, coeur, foie, cortex rénal), particulièrement pendant les périodes interprandiales [1]. La concentration en AGL est augmentée tout au long du nycthémère chez les sujets diabétiques de type 2 (Figure 1) [2], ce qui a conduit à l’idée que l’hyperglycémie chez les diabétiques de type 2 pourraient être en partie liée à l’excès d’AGL circulants. Il y a une quarantaine d’années, Sir Philip Randle et ses collaborateurs ont démontré, à partir d’expériences réalisées in vitro sur le coeur de rat isolé et perfusé, l’existence d’un cycle «glucose-acides gras» [3]. Dans ces expériences, une augmentation de la concentration des AGL dans le milieu de perfusion conduisait à une réduction du captage et de l’utilisation du glucose par le myocarde. Ils avaient ensuite généralisé ces observations au muscle squelettique, et suggéré qu’un excès d’AGL pourrait in vivo contribuer à la diminution de l’utilisation du glucose et au développement du diabète de type 2 [4]. Quelques années plus tard, il a été démontré que les acides gras provenant de l’hydrolyse des triglycérides intracellulaires pourraient être aussi importants pour le cycle «glucose-acides gras» que les acides gras provenant de la circulation sanguine [5]. Enfin, l’oxydation des AGL dans le foie fournit des co-facteurs (ATP, NADH, acétyl-CoA) nécessaires à certaines étapes clés de la néoglucogenèse, conduisant à une production accrue de glucose par le foie [6, 7]. Cet aspect est résumé sur la Figure 2 et ne sera pas abordé plus avant dans cet article. Plus récemment, il a été démontré que les acides gras intervenaient dans la régulation de la sécrétion d’insuline par les cellules β des îlots de Langerhans [8]. La notion d’interrelation entre le métabolisme du glucose et le métabolisme des acides gras a ainsi été étendue au foie et aux cellules β des îlots de Langerhans. Ces dernières années, il est apparu, d’une part, que les mécanismes biochimiques initialement proposés pour expliquer le rôle des AGL dans le cycle «glucose-acides gras» étaient en partie erronés et, d’autre part, que les AGL n’étaient pas seulement des carburants énergétiques de la cellule, mais servaient également de molécules de signalisation. En effet, les AGL contrôlent l’expression d’un certain nombre de gènes et, lorsqu’il sont chroniquement présents en excès dans la cellule, sont à l’origine d’une lipotoxicité [9, 10]. Cet article a pour objectif de faire le point sur les connaissances actuelles quant au rôle des acides gras dans la physiopathologie du diabète de type 2.
Figure 1
Variation des concentrations plasmatiques en acides gras libres (AGL) chez des sujets souffrant d’un diabète de type 2 et chez des sujets témoins.
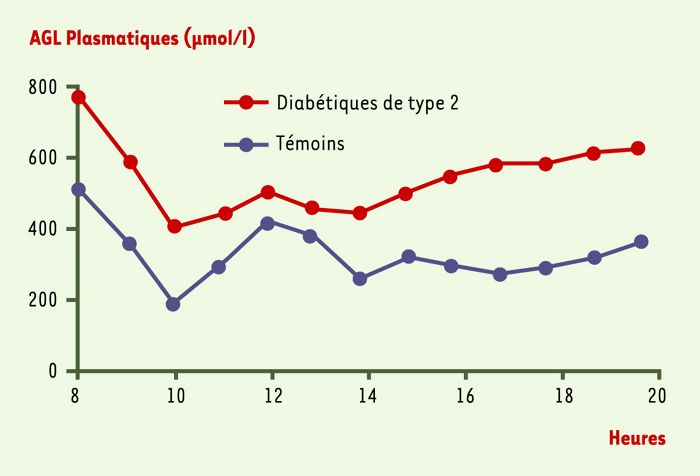
La concentration en AGL est augmentée tout au long du nycthémère chez les sujets diabétiques de type 2, ce qui pourrait être à l’origine des anomalies du métabolisme glucidique chez ces patients (d’après [2]).
Figure 2
Rôle des acides gras dans la régulation de la néoglucogenèse hépatique.

L’oxydation des AGL dans le foie (à droite) fournit des co-facteurs (ATP, NADH, acétyl-CoA) nécessaires à des étapes clés de la néoglucogenèse, conduisant à une production accrue de glucose par le foie (à gauche) (d’après [6]).
Reformulation des mécanismes biochimiques à l’origine du cycle «glucose-acides-gras»
Dans sa description initiale, le cycle «glucose-acides gras» postulait que l’oxydation excessive d’acides gras dans le muscle conduisait à une augmentation des rapports ATP/ADP et acétyl-CoA/CoA, qui inhibent la pyruvate déshydrogénase, l’entrée de l’acétyl-CoA dérivé du pyruvate dans la mitochondrie (Figure 2) et l’oxydation du glucose. Cela reste exact. En revanche, P.J. Randle et al. postulaient dans leur modèle que l’oxydation accrue des acides gras produisait également une augmentation du citrate mitochondrial, en raison de l’afflux d’acétyl-CoA et de l’augmentation de l’ATP et du NADH. Le citrate en excès passait alors dans le cytosol (via un transporteur localisé dans la membrane mitochondriale) où il entraînait une inhibition allostérique de la phosphofructokinase (Figure 3), conduisant à une augmentation de fructose-6-phosphate et de glucose-6-phosphate. L’accumulation de glucose-6-phosphate provoquait une inhibition allostérique de l’hexokinase, conduisant à une augmentation de la concentration intracellulaire en glucose et à l’inhibition de son transport vers l’intérieur de la cellule (Figure 3) (le transport de glucose des muscles est un transport «facilité» dans le sens du gradient de concentration). Il vient en fait d’être établi - en mesurant par résonance magnétique nucléaire la concentration de ces métabolites dans le muscle exposé à des concentrations élevées d’acides gras - que certains des mécanismes proposés par P.J. Randle et al. sont inexacts. En effet, les concentrations de glucose-6-phosphate et de glucose sont en réalité diminuées dans le muscle exposé à des concentrations élevées d’acides gras.
Figure 3
Le cycle «glucose-acides gras» de Randle.

L’accumulation de citrate, en inhibant la phosphofructokinase, entraînerait une augmentation de fructose-6-phosphate et de glucose-6-phosphate (G-6-P). Cette accumulation de glucose-6-phosphate inhiberait l’hexokinase, conduisant à l’augmentation du glucose intracellulaire et à l’inhibition du transport de glucose (le transport de glucose des muscles est un transport «facilité», dans le sens du gradient de concentration).Glut: transporteur de glucose; HK: hexokinase; PKF: phospho-fructo-kinase; PDH: pyruvate déshydrogénase (d’après [3]).
Une nouvelle proposition a alors été faite pour expliquer le cycle de Randle, qui suggère que l’excès d’acides gras perturbe les mécanismes de la transmission du signal insulinique [11] (Figure 4). Selon cette hypothèse, l’augmentation des AGL dans le muscle conduirait à l’accumulation de certains de leurs métabolites, tels que l’acyl-CoA et le diacylglycérol. Ceux-ci activent la protéine kinase C, une sérine/thréonine kinase activée par le diacylglycérol [12], qui phosphoryle le substrat majeur du récepteur de l’insuline (IRS) sur des résidus sérine-thréonine, et inhibe ainsi sa capacité de recrutement et d’activation de la phosphatidylinositol 3-kinase (PI3 kinase) [13, 14]. La voie de transduction du signal insulinique est donc diminuée, ce qui conduit à une réduction du transport de glucose. Le même type de mécanisme existe dans le foie, qui conduit à une inhibition de la glycolyse; la résistance à l’insuline entraîne en effet une inactivation (phosphorylation) des enzymes clés (phosphofructo-2 kinase et pyruvate kinase) de la glycolyse, avec pour corollaire une activation de la néoglucogenèse. Cette hypothèse a été étayée par des études récentes réalisées chez des souris dépourvues de tissu adipeux. Celles-ci sont très insulinorésistantes en raison d’un défaut de signalisation insulinique dans les muscles et dans le foie, et présentent une accumulation de triglycérides dans ces tissus [15]. La transplantation d’un tissu adipeux chez ces souris corrige toutes ces anomalies [16], ce qui suggère que l’insulinorésistance observée dans le diabète de type 2, mais aussi dans l’obésité et les lipodystrophies, a pour origine une altération de la répartition des AGL entre les adipocytes, le foie et les muscles ((→) m/s 1999, n°10, p. 1187 et 2001, n°3, p.381).
Figure 4
La reformulation des mécanismes biochimiques à l’origine du cycle «glucose-acides gras» dans les années 2000.

L’excès d’acides gras libres dans le muscle entraîne une inhibition des mécanismes de la transmission du signal insulinique. L’accumulation de métabolites dérivés de ces acides gras libres (acyl-CoA et diacylglycérol) conduit à une activation de la protéine kinase C (à droite) qui, en phosphorylant un résidu sérine (Ser) du substrat majeur du récepteur de l’insuline (IRS), entraîne une diminution du recrutement de la phosphatidylinositol 3-kinase (PI3 kinase) et donc une réduction du transport de glucose (à gauche) (d’après [11]).
Les acides gras libres sont des substrats normaux des cellules β des ilôts de Langerhans
Les acides gras sont des substrats énergétiques majeurs de la cellule β des îlots de Langerhans [17, 18]; ils stimulent l’insulinosécrétion en présence de concentrations normales ou élevées de glucose sanguin. Les effets insulinosécréteurs des acides gras sont néanmoins faibles par rapport à leur contribution au métabolisme oxydatif des cellules β. Les acides gras utilisés par les cellules β ont plusieurs origines: les acides gras libres circulants, les acides gras contenus dans les lipoprotéines circulantes (les cellules β possèdent des récepteurs des LDL qui leur permettent de capter les lipoprotéines circulantes [19]) et les acides gras contenus dans les triglycérides stockés dans la cellule β.
Des expériences récentes ont montré une dualité des effets des acides gras sur la cellule β: l’exposition à court terme (quelques heures) aux AGL potentialise la sécrétion d’insuline en réponse au glucose, tandis que l’exposition à long terme (plusieurs jours) inhibe la sécrétion d’insuline en réponse au glucose.
In vivo, la sécrétion rapide d’insuline en réponse au glucose est la même chez le rat nourri ou à jeun, alors que la sécrétion tardive est diminuée chez le rat nourri. L’une des différences entre les deux groupes d’animaux est que la concentration en AGL est beaucoup plus élevée chez le rat à jeun [20]. Si l’on injecte un agent antilipolytique (acide nicotinique) au rat à jeun, afin de diminuer la concentration en AGL avant la perfusion de glucose, la sécrétion tardive d’insuline en réponse au glucose est supprimée. Si la concentration en AGL est maintenue par perfusion de triglycérides et d’héparine, la sécrétion d’insuline en réponse au glucose est restaurée. L’effet potentialisateur à court terme des acides gras sur l’insulinosécrétion en réponse au glucose est probablement dû à une augmentation de la concentration d’acyl-CoA dans le cytosol. En effet, ceux-ci stimulent l’exocytose, soit directement, soit après formation de diacylglycérol, un stimulateur de la protéine kinase C (PKC) [8]. Bien qu’il existe une certaine controverse à ce sujet [8], l’exposition prolongée des îlots de Langerhans aux acides gras induit généralement in vivo une diminution de la sécrétion d’insuline en réponse au glucose [21, 22]. En revanche, les études réalisées in vitro montrent clairement que l’exposition prolongée des îlots de Langerhans aux acides gras entraîne une inhibition de la sécrétion tardive d’insuline en réponse au glucose par le pancréas isolé et perfusé [8].
Plusieurs hypothèses ont été avancées pour expliquer ce rôle inhibiteur des acides gras sur l’insulinosécrétion en réponse au glucose. La première est en relation avec le fait que les acyl-CoA stimulent le canal K+ dépendant de l’ATP en se liant à la protéine Kir6.2 qui forme le pore du canal. L’accumulation des acyl-CoA dans la cellule β, en réponse à une exposition chronique à des concentrations élevées d’acides gras, empêcherait ainsi la fermeture du canal K+ dépendant de l’ATP et contribuerait à une perte de sensibilité de la cellule β au glucose. La seconde hypothèse est en relation avec une modification de l’expression de gènes contrôlant le métabolisme du glucose ou des acides gras: l’exposition in vitro d’îlots de Langerhans ou de cellules β à des concentrations élévées d’acides gras entraîne une diminution de l’expression du transporteur de glucose GLUT2, de la glucokinase, du facteur de transcription IDX-1 (islet-duodenum-homeobox-1) et de l’acétyl-CoA carboxylase (et donc du malonyl-CoA) [23], ainsi qu’une augmentation de l’expression de la carnitine palmitoyltransférase I (CPT-I), enzyme contrôlant l’entrée des acides gras à longue chaîne dans la mitochondrie [8] ((→) m/s 1998, n°2, p.206). Cela aboutit à une diminution du métabolisme du glucose dans les îlots et à une augmentation de l’oxydation des acides gras, réduisant ainsi la concentration des acyl-CoA cytoplasmiques. La troisième hypothèse permettant d’expliquer l’inhibition de l’insulinosécrétion par les AGL est en relation avec un effet découplant des acides gras sur la mitochondrie. L’exposition prolongée des îlots de Langerhans aux acides gras entraîne les signes classiques du découplage mitochondrial: augmentation de la respiration, diminution du potentiel membranaire et de la synthèse d’ATP, gonflement de la mitochondrie. Ce découplage est probablement partiellement responsable de l’inhibition de l’insulinosécrétion en réponse au glucose (l’ATP étant nécessaire à la fermeture du canal K+ dépendant de l’ATP). On observe également une augmentation de la production de radicaux libres (ROS, reactive oxygen species) [8], qui a probablement une importance dans les phénomènes d’apoptose décrits plus loin.
Lipotoxicité et étiologie du diabète de type 2
Il existe dans le diabète de type 2 une perte de la première phase de l’insulinosécrétion, une disparition des oscillations sécrétoires, une sécrétion retardée et réduite d’insuline en réponse au repas et une augmentation de la sécrétion de pro-insuline. Les acides gras sont considérés comme des facteurs importants participant à l’étiologie du diabète de type 2 [24, 25].
Le développement du diabète chez le rat ZDF (Zucker diabetic fatty) présente de nombreuses analogies avec le diabète de type 2 observé chez l’homme [26]. Les rats ZDF deviennent obèses en raison d’une mutation (fa) dans le domaine extracellulaire du récepteur de la leptine. À l’état homozygote, ils présentent progressivement une insulinorésistance et une intolérance au glucose entre 3 et 8 semaines, et deviennent véritablement diabétiques entre 8 et 10 semaines ((→) m/s 1999, n°8-9, p.1060). Les rats hétérozygotes ne présentent ni insulinorésistance, ni intolérance au glucose, et ne deviennent jamais diabétiques. L’apparition du diabète s’accompagne d’une hypertrophie des îlots de Langerhans et d’une perte de la sécrétion d’insuline en réponse au glucose [8, 10, 27]. Par la suite, et malgré des capacités prolifératives plus élevées, la masse de cellules β est réduite de 50%, suggérant une augmentation de l’apoptose [28]. Une évolution similaire est observée dans un autre modèle de rat présentant une hypertriglycéridémie et un diabète de type 2, le rat OLETF (Otsuka Long-Evans Tokushima fatty) [29].
Des quantités considérables de triglycérides s’accumulent dans les îlots de Langerhans des rats ZDF au début de la maladie [8, 10]. Puis, les îlots de Langerhans perdent 50% de leurs cellules β par apoptose (mort cellulaire programmée) [8, 10]. La surcharge en lipides résulte d’une augmentation de la concentration des acides gras libres et des triglycérides circulants, couplée à une capacité accrue de lipogenèse (transformation du glucose en acides gras) [8, 10]. L’expression et l’activité des enzymes de la lipogenèse est augmentée, tandis que celles des enzymes de l’oxydation des acides gras est diminuée [23]. L’accumulation de triglycérides dans les îlots de Langerhans est associée à de très nombreuses anomalies: diminution de l’expression du transporteur de glucose GLUT2 et de la sécrétion d’insuline, augmentation de la formation de monoxyde d’azote (NO) et stimulation de l’apoptose. La stimulation de l’apoptose est liée à la réduction du facteur anti-apoptotique Bcl-2, sans modification du facteur apoptotique Bax. Ce phénomène a été appelé «lipotoxicité» [10, 30]) (Figure 5).
Figure 5
Effets cytotoxiques des acides gras sur la cellule β du pancréas endocrine: mécanismes par lesquels la troglitazone prévient la cytotoxicité.
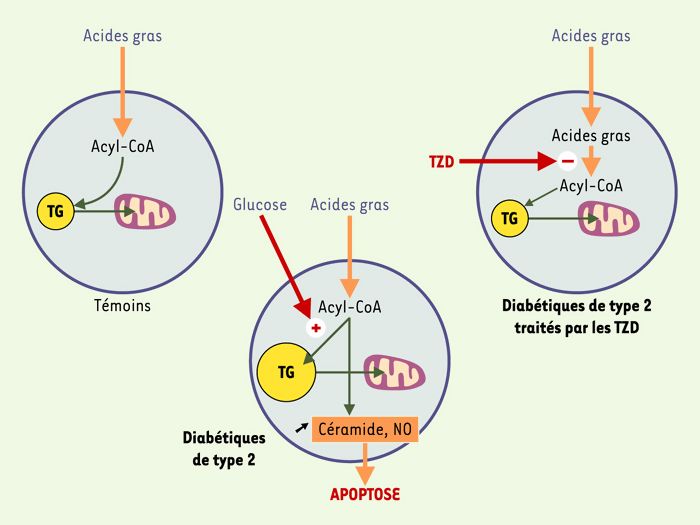
L’accumulation de triglycérides (TG) dans les îlots de Langerhans entraînerait une augmentation de la synthèse de céramide, l’excès de palmitoyl-CoA se condensant à la sérine pour former un céramide. Celui-ci stimulerait la production de monoxyde d’azote (NO), et le déclenchement de l’apoptose. La troglitazone, agent hypoglycémiant, diminue l’expression des enzymes impliquées dans l’activation (acyl-CoA synthétase) et l’estérification (glycérolphosphate acyltransférase) des acides gras, d’où une réduction du contenu en triglycérides des îlots de Langerhans. Cette diminution entraîne, d’une part, une amélioration de l’insulinosécrétion en réponse au glucose et, d’autre part, une réduction des phénomènes apoptotiques des cellules β (d’après [8]).
La chronologie du phénomène de lipotoxicité serait la suivante: le contenu élevé en lipides entraînerait une augmentation de la synthèse de céramide, l’excès de palmitoyl-CoA se condensant à la sérine pour former un céramide (un dérivé de sphingosine et un précurseur des sphingolipides). Le céramide augmenterait ensuite l’expression de la forme inductible de la mono-oxygénase synthase (iNOS), ce qui se traduirait par une surproduction de NO et le déclenchement de l’apoptose. Plusieurs arguments expérimentaux sont en faveur de ce mécanisme: ce scénario peut être reproduit en traitant les îlots de Langerhans de rats ZDF prédiabétiques avec un C2-céramide; les effets apoptotiques sont entièrement bloqués par des inhibiteurs de la synthèse de céramide (fumonisine B1) et de NO (nicotinamide, aminoguanidine). Le NO et le peroxynitrite (produit de réaction de NO et des ions superoxydes) ont pour cible la mitochondrie: le NO, en se fixant au site de liaison de l’oxygène sur la cytochrome C oxydase, est un inhibiteur puissant, mais réversible, de la chaîne respiratoire. On observe ensuite une ouverture du pore de perméabilité de transition (MTP), ce qui entraîne une fuite de protons et l’hydrolyse de l’ATP (découplage des phosphorylations oxydatives). La mitochondrie gonfle et libère du cytochrome C qui active des protéases cytoplasmiques, les caspases, responsables d’une stimulation de la protéolyse et de la destruction des cellules (Figure 6).
Figure 6
Mécanismes biochimiques impliqués dans la lipotoxicité.
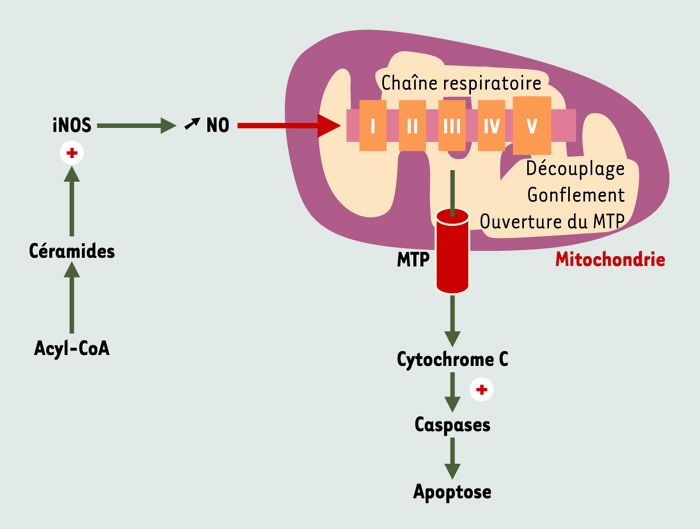
Le monoxyde d’azote (NO) se fixe sur le site de liaison de l’oxygène sur la cytochrome C oxydase, ce qui entraîne une inhibition de la chaîne respiratoire. L’ouverture du pore de perméabilité de transition (MTP) s’accompagne d’un découplage des phosphorylations oxydatives. La mitochondrie gonfle et libère du cytochrome C qui active des protéases cytoplasmiques, les caspases, responsables de l’apoptose des cellules. iNOS: inducible nitric oxide synthase.
La surcharge lipidique des îlots de Langerhans peut être prévenue par la restriction calorique et par l’administration dans le pancréas d’un adénovirus contenant l’ADNc du récepteur de la leptine, permettant ainsi de corriger l’anomalie du récepteur de la leptine, ou par l’administration de troglitazone, un agent antidiabétique qui agit en se liant à un récepteur nucléaire de type PPAR (peroxisome proliferator activated receptor) [8, 10]. L’administration dans le pancréas de l’adénovirus contenant l’ADNc du récepteur de la leptine restaure l’expression du transporteur de glucose GLUT2 et de la glucokinase, ainsi que la sécrétion d’insuline en réponse au glucose. Cette administration restaure également l’expression de Bcl-2 et inhibe l’apoptose [8, 10]. L’hypothèse selon laquelle le principal rôle de la leptine, via son récepteur, serait de contrôler le contenu intracellulaire en triglycérides a été avancée [10, 31]. Le contenu en triglycérides des îlots de Langerhans peut également être réduit en traitant les rats ZDF avec un agent hypoglycémiant, la troglitazone, qui se lie à des récepteurs nucléaires. Le traitement des rats ZDF par la troglitazone diminue l’expression des enzymes impliquées dans l’activation (acyl-CoA synthétase) et l’estérification (glycérolphosphate acyltransférase) des acides gras, réduit le contenu des îlots en triglycérides, améliore l’insulinosécrétion en réponse au glucose [32] et prévient l’apoptose [33] (Figure5). Cet effet lipostatique de la troglitazone en fait un agent pharmacologique potentiellement intéressant pour ralentir, voire prévenir, le passage des diabétiques de type 2 d’un état d’insulinorésistance compensé à un état d’insulinorequérance.
Conclusions
Il est clairement établi que les acides gras sont des substrats énergétiques des cellules β des îlots de Langerhans, et que les acides gras à longue chaîne potentialisent à court terme la sécrétion d’insuline en réponse au glucose. Les mécanismes cellulaires de cette potentialisation ne sont pas entièrement élucidés, mais il semble que l’accumulation des acyl-CoA dans le cytosol conduit à la production de diacylglycérol et de phospholipides qui stimulent l’exocytose des granules d’insuline [27].
Lorsque les îlots de Langerhans sont exposés de manière chronique à des concentrations élevées d’acides gras, comme c’est le cas chez les sujets présentant un diabète de type 2, des triglycérides s’accumulent dans les cellules β et produisent une inhibition de la sécrétion d’insuline en réponse au glucose ainsi qu’une lipotoxicité qui conduit à la destruction des cellules par apoptose. À l’examen anatomopathologique, la présence de vésicules lipidiques à l’intérieur des cellules β des îlots a été observée chez des diabétiques de type 2, suggérant que la lipotoxicité pourrait rendre compte de la disparition des cellules β observée chez les diabétiques de type 2 passant d’un état d’insulinorésistance compensé à un état d’insulinorequérance.
Lorsque les îlots de Langerhans sont exposés de manière chronique à des concentrations élevées de glucose, un phénomène de glucotoxicité a également été décrit [34]. La part relative de la glucotoxicité et de la lipotoxicité dans la perte des cellules β n’a pas été établie, mais il est clair que ces deux mécanismes doivent être impliqués chez les diabétiques de type 2 exposés de façon chronique à des concentrations élevées de glucose et d’acides gras [35].
Appendices
Références
- 1. Owen OE, Reichard GA, Patel MS, Boden G. Energy metabolism in feasting and fasting. In: Klachko DM, Anderson RR, Heimberg M, eds. Hormone and energy metabolism. New York: Plenum Press, 1979: 169-88.
- 2. Reaven GM, Hollenbeck CB, Jen CY, Wu MS, Chen YDI. Measurement of plasma glucose, free fatty acid, lactate and insulin for 24 h in patients with NIDDM. Diabetes 1988; 37: 1020-4.
- 3. Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose-fatty acid cycle: its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963; I: 785-9.
- 4. Randle PJ, Kerbey AL, Espinal J. Mechanisms decreasing glucose oxidation in diabetes and starvation: role of lipid fuels and hormones. Diabetes Metab Rev 1988; 4: 623-8.
- 5. Zierler KL. Fatty acids as substrates for heart and skeletal muscle. Circ Res 1976; 39: 459-63.
- 6. Girard J. Rôle des acides gras libres dans l’insulino-résistance au cours du diabète non-insulino-dépendant. Diabete Metab 1995; 21: 79-88.
- 7. Boden G. Effects of free fatty acids on gluconeogenesis and glycogenolysis. Life Sci 2003; 72: 977-88.
- 8. Girard J. Acides gras, insulinosécrétion et lipotoxicité. Med Ther Endocrinol 2000; 2 (suppl2): 29-36.
- 9. Unger RH. Lipotoxicity in the pathogenesis of obesity-dependent NIDDM: genetic and clinical implications. Diabetes 1995; 44: 863-70.
- 10. Unger RH, The physiology of cellular liporegulation. Annu Rev Physiol 2003; 65: 333-47.
- 11. Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest 2000; 106: 171-6.
- 12. Griffin ME, Marcucci MJ, Cline GW, et al. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 1999; 48: 1270-4.
- 13. Roden M, Price TB, Perseghin G, et al. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest 1996; 97: 2859-2865.
- 14. Dresner A, Laurent D, Marcucci M, et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest 1999; 103: 253-9.
- 15. Kim JK, Gavrilova O, Chen Y, Reitman ML, Shulman GI. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J Biol Chem 2000; 275: 8456-60.
- 16. Gavrilova O, Marcus-Samuels B, Graham D, et al. Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J Clin Invest 2000; 105: 271-8.
- 17. Berne C. The metabolism of lipids in mouse pancreatic islets: the oxidation of fatty acids and ketone bodies. Biochem J 1975; 152: 661-6.
- 18. Malaisse WJ, Malaisse-Lagae F, Sener A, Hellerström C. Participation of endogenous fatty acids in the secretory activity of the pancreatic β-cell. Biochem J 1985; 227: 995-1002.
- 19. Grupping AY, Cnop M, Van Schravendijk C, Hannaert J, Van Berkel T, Pipeleers D. Low density lipoprotein binding and uptake by human and rat islet β-cells. Endocrinology 1997; 138: 4064-8.
- 20. Stein DT, Esser V, Stevenson BE, et al. Essentiality of circulating fatty acids for glucose-stimulated insulin secretion in the fasted rat. J Clin Invest 1996; 97: 2728-35.
- 21. Mason TM, Goh T, Tchipashvili V, et al. Prolonged elevation of plasma free fatty acids desensitizes the insulin secretory response to glucose in vivo in rats. Diabetes 1999; 48: 524-30.
- 22. Paolisso G, Gambardella A, Amato L, et al. Opposite effects of short- and long-term fatty acid infusion on insulin secretion in healthy subjects. Diabetologia 1995; 38: 1295-9.
- 23. Prentki M, Joly E, El-Assaad W, Roduit R. Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity: role in β-cell adaptation and failure in the etiology of diabetes. Diabetes 2002; 51 (suppl3): S405-13.
- 24. McGarry JD. What if Minkowski had been ageusic ? An alternative angle on diabetes. Science 1992; 258: 766-70.
- 25. McGarry JD. Dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 2002; 51: 7-18.
- 26. Unger RH. How obesity causes diabetes in Zucker diabetic fatty rats. Trends Endocrinol Metab 1997; 8: 276-82.
- 27. McGarry JD, Dobbins RL. Fatty acids, lipotoxicity and insulin secretion. Diabetologia 1999; 42: 128-38.
- 28. Pick A, Clark J, Kubstrup C, et al. Role of apoptosis in failure of β-cell mass compensation for insulin resistance and β-cell defects in the male Zucker diabetic fatty rat. Diabetes 1998; 47: 358-64.
- 29. Man ZW, Zhu M, Noma Y, et al. Impaired β-cell function and deposition of fat droplets in the pancreas as a consequence of hypertriglyceridemia in OLETF rat, a model of spontaneous NIDDM. Diabetes 1997; 46: 1718-24.
- 30. Unger RH. Lipotoxic diseases. Annu Rev Med 2002; 53: 319-36.
- 31. Unger RH, Zhou YT, Orci L. Regulation of fatty acid homeostasis in cells: novel role of leptin. Proc Natl Acad Sci USA 1999; 96: 2327-32.
- 32. Shimabukuro M, Zhou YT, Lee Y, Unger RH. Troglitazone lowers islet fat and restores β-cell function of Zucker diabetic fatty rats. J Biol Chem 1998; 273: 3547-50.
- 33. Higa M, Zhou YT, Ravazzola M, Baetens D, Orci L, Unger RH. Troglitazone prevents mitochondrial alterations, β-cell destruction, and diabetes in obese prediabetic rats. Proc Natl Acad Sci USA 1999; 96: 11513-8.
- 34. Marshak S, Leibowitz G, Bertuzzi F, et al. Impaired β-cell functions induced by chronic exposure of cultured human pancreatic islets to high glucose. Diabetes 1999; 48: 1230-6.
- 35. Poitout V, Robertson RP. Secondary β-cell failure in type 2 diabetes. A convergence of glucotoxicity and lipotoxicity. Endocrinology 2002; 143: 339-42.
List of figures
Figure 1
Variation des concentrations plasmatiques en acides gras libres (AGL) chez des sujets souffrant d’un diabète de type 2 et chez des sujets témoins.
La concentration en AGL est augmentée tout au long du nycthémère chez les sujets diabétiques de type 2, ce qui pourrait être à l’origine des anomalies du métabolisme glucidique chez ces patients (d’après [2]).
Figure 2
Rôle des acides gras dans la régulation de la néoglucogenèse hépatique.
L’oxydation des AGL dans le foie (à droite) fournit des co-facteurs (ATP, NADH, acétyl-CoA) nécessaires à des étapes clés de la néoglucogenèse, conduisant à une production accrue de glucose par le foie (à gauche) (d’après [6]).
Figure 3
Le cycle «glucose-acides gras» de Randle.
L’accumulation de citrate, en inhibant la phosphofructokinase, entraînerait une augmentation de fructose-6-phosphate et de glucose-6-phosphate (G-6-P). Cette accumulation de glucose-6-phosphate inhiberait l’hexokinase, conduisant à l’augmentation du glucose intracellulaire et à l’inhibition du transport de glucose (le transport de glucose des muscles est un transport «facilité», dans le sens du gradient de concentration).Glut: transporteur de glucose; HK: hexokinase; PKF: phospho-fructo-kinase; PDH: pyruvate déshydrogénase (d’après [3]).
Figure 4
La reformulation des mécanismes biochimiques à l’origine du cycle «glucose-acides gras» dans les années 2000.
L’excès d’acides gras libres dans le muscle entraîne une inhibition des mécanismes de la transmission du signal insulinique. L’accumulation de métabolites dérivés de ces acides gras libres (acyl-CoA et diacylglycérol) conduit à une activation de la protéine kinase C (à droite) qui, en phosphorylant un résidu sérine (Ser) du substrat majeur du récepteur de l’insuline (IRS), entraîne une diminution du recrutement de la phosphatidylinositol 3-kinase (PI3 kinase) et donc une réduction du transport de glucose (à gauche) (d’après [11]).
Figure 5
Effets cytotoxiques des acides gras sur la cellule β du pancréas endocrine: mécanismes par lesquels la troglitazone prévient la cytotoxicité.
L’accumulation de triglycérides (TG) dans les îlots de Langerhans entraînerait une augmentation de la synthèse de céramide, l’excès de palmitoyl-CoA se condensant à la sérine pour former un céramide. Celui-ci stimulerait la production de monoxyde d’azote (NO), et le déclenchement de l’apoptose. La troglitazone, agent hypoglycémiant, diminue l’expression des enzymes impliquées dans l’activation (acyl-CoA synthétase) et l’estérification (glycérolphosphate acyltransférase) des acides gras, d’où une réduction du contenu en triglycérides des îlots de Langerhans. Cette diminution entraîne, d’une part, une amélioration de l’insulinosécrétion en réponse au glucose et, d’autre part, une réduction des phénomènes apoptotiques des cellules β (d’après [8]).
Figure 6
Mécanismes biochimiques impliqués dans la lipotoxicité.
Le monoxyde d’azote (NO) se fixe sur le site de liaison de l’oxygène sur la cytochrome C oxydase, ce qui entraîne une inhibition de la chaîne respiratoire. L’ouverture du pore de perméabilité de transition (MTP) s’accompagne d’un découplage des phosphorylations oxydatives. La mitochondrie gonfle et libère du cytochrome C qui active des protéases cytoplasmiques, les caspases, responsables de l’apoptose des cellules. iNOS: inducible nitric oxide synthase.