Abstracts
Résumé
La chorée de Huntington est une maladie neurodégénérative héréditaire dominante, caractérisée par l’apparition progressive d’une dyskinésie, de déficits cognitifs et de troubles émotionnels. Près d’une décennie après l’identification du gène et de la mutation responsable de cette maladie, celle-ci reste incurable. Néanmoins, le développement de modèles transgéniques a permis une avancée majeure dans la connaissance des mécanismes cellulaires et moléculaires précoces de la maladie. La mutation conduirait à un dérèglement de la transcription, à une altération de la dégradation des protéines défectueuses par les protéasomes, ainsi qu’à des processus excitotoxiques et à un dysfonctionnement des mitochondries. Cet article souligne les apports récents de l’utilisation de modèles transgéniques chez la souris et chez la drosophile dans la compréhension de la pathogénie et dans l’élaboration de nouvelles stratégies thérapeutiques.
Summary
Huntington’s disease is an hereditary dominant neurodegenerative disorder clinically characterised by progressive dyskinesia, cognitive decline and psychiatric disturbances. One decade after the identification of the gene whose mutation is responsible for the disease, this pathology remains incurable. However, major insights into early cellular and molecular basis of Huntington’s disease have arisen from transgenic models. Transcriptional dysregulation, abnormal degradation of misfolded proteins as well as excitotoxic processes and mitochondrial dysfunction are involved in Huntington’s disease. The present review discusses the recent insights gained from mouse and Drosophila models towards the understanding of pathogenesis and the development of new therapeutic tools.
Article body
La chorée de Huntington est une maladie neurodégénérative dévastatrice, dont souffre environ 1 individu sur 10000 et qui apparaît généralement entre 35 et 45 ans. Elle peut néanmoins survenir à d’autres périodes de la vie, dès l’enfance ou après la 5e décennie. Les patients atteints de cette maladie présentent des désordres moteurs caractérisés principalement par des mouvements brusques involontaires (chorea signifie danse en grec) ((→) m/s 1997, n° 6-7, p. 850), une ataxie, une bradykinésie et une dystonie musculaire. La maladie évolue avec l’intensification des déficits moteurs, l’apparition progressive de troubles émotionnels, voire d’un état de démence, et débouche irrémédiablement sur la mort après 15 à 20 ans d’évolution ((→) m/s 1988, n° 8, p. 492).
Sur le plan histologique, les patients présentent une dégénérescence progressive des neurones du striatum (noyau caudé et putamen) et, de façon moindre, des neurones du cortex cérébral et du cervelet, associée à une réaction astrocytaire. La chorée de Huntington est une maladie héréditaire autosomique dominante. Elle a pour origine une répétition anormale de codons CAG (36 à 180 répétitions) dans l’exon 1 du gène IT15 codant pour la protéine huntingtine [1]. Cette séquence répétée est traduite en une longue chaîne polyglutamine (polyQ) dans la région amino-terminale de la protéine. Au moins huit autres neuropathologies résulteraient de la répétition anormale de codons CAG dans certains gènes: les ataxies spino-cérébelleuses de type 1, 2, 3, 6, 7 et 12, l’amyotrophie spino-bulbaire ainsi que l’atrophie dentato-rubro-pallido-luysienne. Toutes ces maladies présentent comme point commun la présence d’agrégats dans le noyau des neurones (inclusion intranucléaire).
À ce jour, ni la fonction exacte de la huntingtine, ni le rôle joué par les inclusions intranucléaires ne sont clairement élucidés. La huntingtine serait impliquée dans le trafic vésiculaire et aurait des propriétés anti-apoptotiques. Quant aux inclusions intranucléaires, leur rôle dans la maladie est partagé entre deux hypothèses contraires, un effet neuroprotecteur ou un effet délétère. Le développement récent de modèles transgéniques de la chorée de Huntington, en particulier chez la souris et chez la drosophile ((→) m/s 2000, n° 2, p. 164), a permis une meilleure compréhension de la pathogénie et a provoqué l’émergence de nouvelles stratégies thérapeutiques.
Les modèles chez la souris
Les lignées R6
Les premiers modèles transgéniques chez la souris ont été mis au point par le groupe de Bates en 1996 sous le nom de R6 [2]. L’interruption de l’expression du gène IT15 étant létale au stade embryonnaire chez la souris, il a très tôt été suggéré que la mutation à l’origine de la chorée de Huntington devait entraîner un gain de fonction plutôt qu’une perte. Sur cette base, l’introduction d’une seule copie du gène IT15 humain muté dans le génome murin devait être suffisante pour produire une maladie. Les lignées R6 (R6/0, R6/1, R6/2 et R6/5) ont été établies après insertion de l’exon 1 du gène IT15 humain avec une longue répétition (> 115) de CAG.
La lignée murine R6/2 est de loin la plus étudiée et la mieux caractérisée. Les souris R6/2 présentent de façon précoce des déficits moteurs et d’apprentissage qui augmentent avec l’âge, ainsi qu’une baisse progressive de leur poids [2]. Les animaux meurent prématurément, mais ne montrent pas de dégénérescence neuronale massive [2, 3]. Cette dernière observation a suggéré que les troubles comportementaux proviendraient non pas d’une neurodégénérescence, mais plutôt d’un dysfonctionnement précoce des circuits neuronaux. De nombreuses données corroborent maintenant cette hypothèse (voir ci-dessous).
Très tôt après le développement des lignées R6, l’utilisation d’anticorps dirigés contre la partie amino-terminale de la huntingtine mutée, c’est-à-dire contre le fragment correspondant au transgène, a révélé l’existence d’inclusions intranucléaires, mais aussi d’agrégats cytoplasmiques contenant cette protéine (Figure 1) dans les neurones. Outre le système nerveux, d’autres organes tels que les muscles squelettiques, le coeur et le foie présentent des inclusions. Celles-ci ont également été retrouvées dans le cerveau de patients atteints de chorée de Huntington.
La découverte de ces inclusions a dès lors bouleversé la recherche sur cette neuropathologie, d’autant que leur formation précède l’apparition des déficits comportementaux dans les lignées R6. Ces inclusions peuvent donc servir de marqueurs précoces de la maladie, et il a été proposé qu’elles pouvaient jouer un rôle direct dans la pathogénie (Tableau I).
Tableau I
Modèles de souris transgéniques utilisés pour l’analyse de la chorée de Huntington.

Tableau I (continuation)
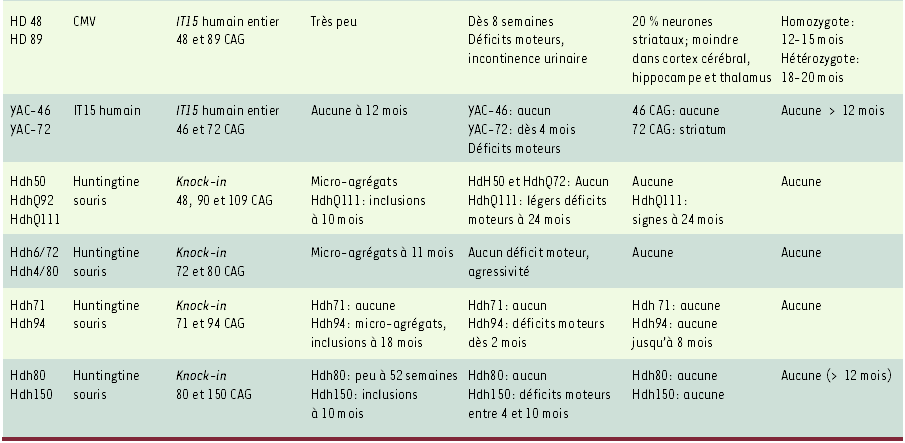
IT15: gène humain codant pour la huntingtine. CMV: cytomégalovirus. Les résultats obtenus avec ces modèles murins suggèrent que les déficits comportementaux observés dans la chorée de Huntington précèdent l’apparition des dégénérescences neuronales et sont probablement causés par un dysfonctionnement cellulaire. Aucune corrélation claire n’existe entre la mort neuronale et la mortalité des animaux, ni avec l’apparition des inclusions [2-8, 20, 27-30].
Figure 1
Inclusions intranucléaires et agrégats extranucléaires dans le striatum d’une souris transgénique R6/2.
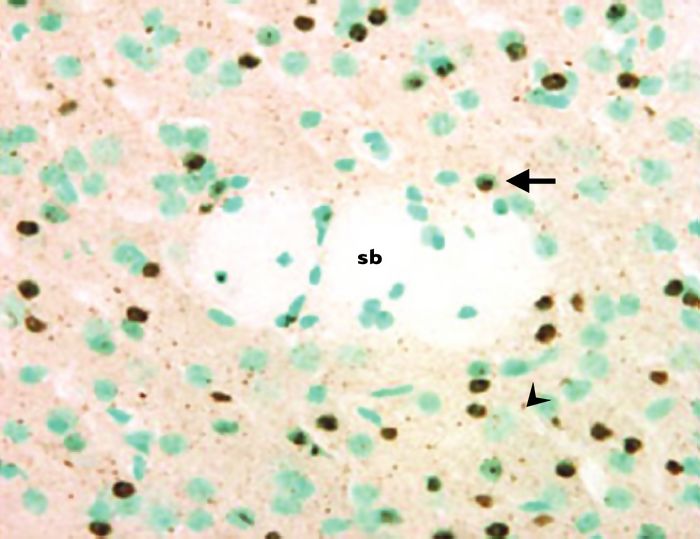
Ce marquage a été obtenu en utilisant un anticorps reconnaissant la partie amino-terminale de la huntingtine mutée. Les noyaux cellulaires sont colorés au vert de méthyle. La flèche indique une inclusion intranucléaire, et la pointe de flèche un agrégat extranucléaire. Ces deux types d’agrégats sont également présents dans le cerveau de patients atteints de chorée de Huntington. sb: substance blanche correspondant à la capsule interne traversant le striatum (photo, J.C. Liévens).
Les lignées exprimant le gène IT15 humain entier
Deux autres séries de lignées transgéniques ont depuis été développées avec l’ADNc complet du gène IT15 humain muté. Reddy et al. [4] ont ainsi montré que l’expression de la protéine huntingtine entière mutée induisait des dégénérescences neuronales partielles et des déficits comportementaux chez la souris. De même, l’introduction d’un chromosome artificiel de levure contenant un tel ADNc complet (lignées YAC) entraîne la dégénérescence sélective des neurones du striatum [5] (Tableau I).
Les lignées knock-in
Ces lignées ont été obtenues en mutant le gène murin codant pour la huntingtine afin d’y insérer des répétitions du codon CAG. Ces souris ne montrent des déficits comportementaux que très tardivement [6-8] et, à ce jour, des signes de mort neuronale n’ont été observés que dans l’une de ces lignées [8]. Néanmoins, ces souris présenteraient certaines altérations moléculaires similaires à celles observées dans les lignées R6, notamment en ce qui concerne la présence d’inclusions intranucléaires [6-8] (Tableau I).
Les modèles chez la drosophile
L’introduction d’organismes simples tels que la drosophile et C. elegans comme modèles d’étude de la chorée de Huntington a permis une accélération des recherches dans ce domaine ((→) m/s 2000, n° 2, p. 164). Les modèles développés chez la drosophile présentent des caractéristiques pathologiques comparables à celles observées dans la maladie humaine, entre autres une dégénérescence neuronale progressive, qui ne se retrouve pas dans la plupart des modèles murins, et la formation d’agrégats intracellulaires par la protéine huntingtine mutée [9, 10]. Par ailleurs, puisque la drosophile et C. elegans ont une durée de génération très courte et peuvent être maintenus à moindre frais en très grand nombre, leur utilisation montre un net avantage, par rapport aux modèles mammifères, pour tester rapidement divers principes actifs à visée thérapeutique. Enfin, ces organismes ont fait l’objet d’études génétiques pionnières depuis de longues années, permettant ainsi d’accéder à un large éventail d’approches génétiques.
Deux groupes ont mis au point des modèles de la chorée de Huntington chez la drosophile. Ces modèles consistent à exprimer un fragment amino-terminal du gène huntingtine humain contenant une répétition élevée du codon CAG, sous le contrôle d’un promoteur neuronal ou spécifique de la rétine [9, 11]. D’autres auteurs ont simplement exprimé chez la drosophile une longue séquence répétée de CAG [12, 13]. Dans ce dernier cas, le modèle peut être considéré plus généralement comme un modèle des maladies à expansion de polyQ. L’expression de ces différents transgènes dans la rétine de drosophile a entraîné une dégénérescence progressive des photorécepteurs.
Les mécanismes moléculaires et cellulaires de la pathogénie
Interaction protéine-protéine
Une des questions primordiales dans la compréhension de la chorée de Huntington est celle du rôle joué par les agrégats de huntingtine mutée. Les données sur ce sujet restent encore controversées et ne peuvent être détaillées ici: certains auteurs proposent que la formation d’agrégats pourrait représenter un mécanisme neuroprotecteur, tandis que d’autres suggèrent que les agrégats seraient cytotoxiques. L’étude récente menée par Kazantsev et al. [10] chez la drosophile a apporté un nouvel argument en faveur de la cytotoxicité, en montrant que l’expression de peptides suppresseurs de l’agrégation de la protéine huntingtine mutée prévient la mort neuronale ainsi que la forte létalité observées dans ces modèles ((→) m/s 2000, n° 1, p. 57). Cependant, il est possible que les agrégats de huntingtine ne jouent pas un rôle essentiel dans l’induction des processus dégénératifs: ainsi, alors qu’une perte neuronale est détectée dans les lignées transgéniques exprimant le gène IT15 muté entier, la formation d’inclusions intranucléaires y est très réduite, voire quasi absente [4, 5].
L’interaction de la huntingtine mutée avec d’autres molécules, avant qu’elle ne forme des agrégats, pourrait priver la cellule de protéines essentielles à son fonctionnement et à sa survie. Deux mécanismes hypothétiques pour l’interaction anormale de la huntingtine mutée avec elle-même ou avec d’autres protéines ont été décrits dans la littérature (Figure 2). Perutz et al. [14] ont proposé que les longues chaînes polyQ présentes dans un peptide aient tendance à s’assembler en feuillet β par des liaisons hydrogène (conformation polar zipper), ce qui entraînerait la formation d’agrégats. Cependant, une telle hypothèse ne permet pas de rendre compte de la lenteur de la cinétique de formation des inclusions chez les patients. Green [15] avait, quant à lui, proposé un mécanisme selon lequel l’interaction entre chaînes polyQ et protéines pourrait s’effectuer grâce à l’intervention d’enzymes telles que les transglutaminases. Ces enzymes catalysent notamment la formation d’une liaison covalente entre la partie carboxamide d’un résidu glutamine et le groupe ε-amine d’un résidu lysine. Diverses études s’accordent à montrer que les chaînes polyQ sont de bons substrats pour l’isoforme tissulaire de la transglutaminase (tTGase, la plus répandue chez les mammifères), et que la réaction est d’autant plus efficace que le nombre de résidus glutamine est important [16]. De plus, l’expression de la tTGase et l’activité transglutaminase sont augmentées dans le cerveau de patients atteints de chorée de Huntington étudiés en post-mortem [16]. L’inhibition de la transglutaminase est donc susceptible de constituer une stratégie thérapeutique. Ainsi, l’administration à des souris transgéniques de cystamine, un inhibiteur de la transglutaminase, permet d’allonger leur survie; mais la cystamine pourrait également avoir un effet neuroprotecteur en inhibant l’activité de caspases impliquées dans la mort cellulaire programmée. Plus récemment, le croisement de souris de la lignée R6/1 avec des souris déficientes en tTGase a permis d’obtenir une diminution sensible de la progression de la maladie; celle-ci était toutefois, curieusement, associée à une formation accrue d’inclusions intranucléaires [17]. Des recherches supplémentaires sont donc nécessaires afin d’élucider le rôle de la tTGase dans cette maladie.
Figure 2
Mécanismes susceptibles de conduire au dysfonctionnement et à la mort des neurones dans la chorée de Huntington.
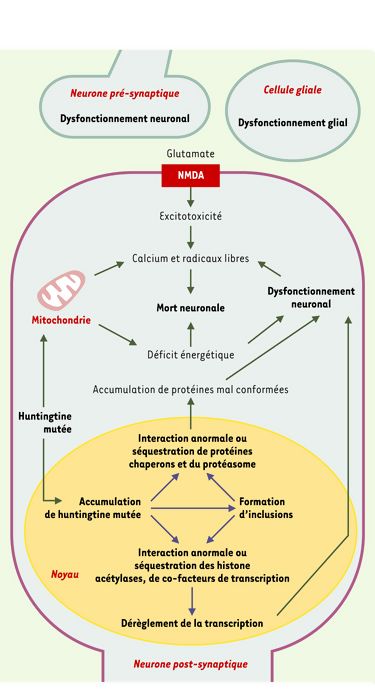
La présence d’une expansion de polyglutamine dans la huntingtine mutée entraîne son accumulation dans le noyau des neurones et la formation d’inclusions intranucléaires. Des protéines chaperons et des constituants du protéasome, d’une part, et des histone acétylases et des co-facteurs de transcription, d’autre part, seraient séquestrés dans ces inclusions ou interagiraient avec la partie amino-terminale de la huntingtine mutée. Cela aurait pour conséquence une accumulation de protéines mal conformées et un dérèglement de la transcription. De plus, la huntingtine mutée affecterait les fonctions mitochondriales. L’ensemble de ces altérations provoquerait un dysfonctionnement des cellules nerveuses. Selon une hypothèse, pour certains types de neurones comme les neurones efférents du striatum, un tel dysfonctionnement, affectant à la fois ces cellules, les neurones pré-synaptiques et les cellules gliales avoisinantes, entraînerait une augmentation des concentrations cytosoliques de calcium et de radicaux libres, notamment au travers de processus excitotoxiques. Ces effets pourraient être à l’origine de la mort neuronale.
Dérèglement de la transcription
La huntingtine mutée pourrait interagir directement avec la machinerie transcriptionnelle en séquestrant des activateurs ou des co-activateurs de transcription tels que CBP (CREB binding protein) [18], ou en s’associant au domaine acétyltransférase des histone acétylases telles que CBP/p300 [9], interférant ainsi avec l’acétylation des histones indispensable à la décondensation de l’ADN. Cette dernière hypothèse est confortée par des travaux princeps, réalisés chez la drosophile, montrant qe l’administration d’inhibiteurs spécifiques des histone désacétylases arrête la progression des neurodégénérescences [9].
Interaction avec les protéines chaperons et le protéasome
Les protéines chaperons permettent aux protéines nouvellement synthétisées d’adopter une conformation fonctionnelle et, à l’inverse, facilitent la dégradation par le protéasome des protéines mal conformées ((→) m/s 2000, n° 5, p. 630). L’un des mécanismes permettant au protéasome d’identifier les protéines à éliminer est la conjugaison de ces protéines à l’ubiquitine. La protéine huntingtine mutée est ubiquitinylée [3] et interagit avec des protéines chaperons de la famille Hsp40/70 [19]. Une hypothèse serait que le protéasome et les protéines chaperons soient submergés par l’accumulation de molécules de huntingtine mal conformées, et ne remplissent plus leur rôle. En accord avec cette idée, il a été observé, par l’intermédiaire d’un criblage génétique, que la surexpression des gènes dHDJ1 ou dTPR2, qui codent tous deux pour des protéines de type chaperon, protège contre les effets dégénératifs dans un modèle de drosophile exprimant une chaîne polyQ seule [12]. Cela reste à confirmer dans les modèles transgéniques de la chorée de Huntington.
Excitotoxicité et dysfonctionnement mitochondrial
L’hypothèse d’une implication de processus excitotoxiques (mort neuronale induite par l’activation excessive des récepteurs du glutamate) dans la chorée de Huntington est depuis longtemps avancée. Des résultats obtenus avec les modèles murins sont venus la corroborer. D’une part, des données électrophysiologiques suggèrent que les récepteurs du glutamate de type NMDA montreraient une hypersensibilité chez les souris transgéniques. D’autre part, des modifications touchant des protéines présynaptiques contrôlant la libération des neurotransmetteurs [21, 22] et une diminution de l’expression d’un transporteur glial de recapture du glutamate [23] ont été décrites. Très récemment, l’administration dans un modèle murin de la chorée de Huntington du riluzole, un agent anti-excitotoxique, ou de rémacémide, un antagoniste des récepteurs NMDA, a permis de réduire la progression de la maladie mais aussi, de façon inattendue, la formation des inclusions intranucléaires [24]. Le riluzole a été testé chez l’homme et donne des résultats prometteurs [25]. Cependant, les mécanismes d’action du riluzole restent à déterminer.
Une altération de l’activité des mitochondries a été très largement démontrée chez les patients atteints de chorée de Huntington [26]. Un déficit du métabolisme énergétique pourrait contribuer, à long terme, à l’induction de dommages neuronaux mettant en jeu notamment des processus excitotoxiques et un stress oxydatif. Les souris transgéniques R6/2 montrent un dysfonctionnement des mitochondries et du métabolisme énergétique. Enfin, l’administration de composés capables d’activer le métabolisme énergétique a des effets bénéfiques, comme par exemple la créatine ou le co-enzyme Q10 associé ou non au rémacémide (antagoniste des récepteurs NMDA) sur des lignées transgéniques murines [24].
Conclusions
L’établissement de modèles transgéniques de la chorée de Huntington a indiscutablement permis d’en étudier la pathogénie, notamment en ce qui concerne les événements précoces. À partir de ces données, des pistes thérapeutiques sont déjà ouvertes, fondées sur une inhibition de l’interaction huntingtine mutée-protéine ou de la désacétylation des histones, ainsi que sur le rôle protecteur des protéines chaperons et des agents anti-excitotoxicité. L’utilisation de modèles chez la drosophile ou chez C. elegans devrait permettre la réalisation de criblages génétiques et l’identification de nouvelles cibles thérapeutiques. L’un des défis sera de déterminer dans l’avenir la nature des mécanismes influençant directement l’évolution de la maladie et permettant d’expliquer sa sélectivité régionale et cellulaire.
Appendices
Remerciements
J.C. Liévens bénéficie d’une aide au retour niveau 1 de la Fondation pour la Recherche Médicale.
Références
- 1. The Huntington’s disease collaborative research group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993; 72: 971-83.
- 2. Mangiarini L, Sathasivam K, Seller M, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996; 87: 493-506.
- 3. Davies SW, Turmaine M, Cozens BA, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997; 90: 537-48.
- 4. Reddy PH, Williams M, Charles V, et al. Behavioural abnormalities and selective neuronal loss in HD transgenic mice expressing mutated full-length HD cDNA. Nat Genet 1998; 20: 198-202.
- 5. Hodgson JG, Agopyan N, Gutekunst CA, et al. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron 1999; 23: 181-92.
- 6. Lin CH, Tallaksen-Greene S, Chien WM, et al. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum Mol Genet 2001; 10: 137-44.
- 7. Menalled LB, Sison JD, Wu Y, et al. Early motor dysfunction and striosomal distribution of huntingtin microaggregates in Huntington’s disease knock-in mice. J Neurosci 2002; 22: 8266-76.
- 8. Wheeler VC, Gutekunst CA, Vrbanac V, et al. Early phenotypes that presage late-onset neurodegenerative disease allow testing of modifiers in Hdh CAG knock-in mice. Hum Mol Genet 2002; 11: 633-40.
- 9. Steffan JS, Bodai L, Pallos J, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 2001; 413: 739-43.
- 10. Kazantsev A, Walker HA, Slepko N, et al. A bivalent Huntingtin binding peptide suppresses polyglutamine aggregation and pathogenesis in Drosophila. Nat Genet 2002; 30: 367-76.
- 11. Jackson GR, Salecker I, Dong X, et al. Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron 1998; 21: 633-42.
- 12. Kazemi-Esfarjani P, Benzer S. Genetic suppression of polyglutamine toxicity in Drosophila. Science 2000; 287: 1837-40.
- 13. Marsh JL, Walker H, Theisen H, et al. Expanded polyglutamine peptides alone are intrinsically cytotoxic and cause neurodegeneration in Drosophila. Hum Mol Genet 2000; 9:13-25.
- 14. Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci USA 1994; 91: 5355-8.
- 15. Green H. Human genetic diseases due to codon reiteration: relationship to an evolutionary mechanism. Cell 1993; 74: 955-6.
- 16. Cooper AJ, Jeitner TM, Gentile V, Blass JP. Cross linking of polyglutamine domains catalyzed by tissue transglutaminase is greatly favored with pathological-length repeats: does transglutaminase activity play a role in (CAG)(n)/Q(n)-expansion diseases? Neurochem Int 2002; 40: 53-67.
- 17. Mastroberardino PG, Iannicola C, Nardacci R, et al. Tissue transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington’s disease. Cell Death Differ 2002; 9: 873-80.
- 18. McCampbell A, Taylor JP, Taye AA, et al. CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet 2000; 9: 2197-202.
- 19. Jana NR, Tanaka M, Wang G, Nukina N. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum Mol Genet 2000; 9: 2009-18.
- 20. Levine MS, Klapstein GJ, Koppel A, et al. Enhanced sensitivity to N-methyl-D-aspartate receptor activation in transgenic and knock-in mouse models of Huntington’s disease. J Neurosci Res 1999; 58: 515-32.
- 21. Cha JH. Transcriptional dysregulation in Huntington’s disease. Trends Neurosci 2000; 23: 387-92.
- 22. Liévens J, Woodman B, Mahal A, Bates G. Abnormal phosphorylation of synapsin I predicts a neuronal transmission impairment in the R6/2 Huntington’s disease transgenic mice. Mol Cell Neurosci 2002; 20: 638-48.
- 23. Liévens JC, Woodman B, Mahal A, et al. Impaired glutamate uptake in the R6 Huntington’s disease transgenic mice. Neurobiol Dis 2001; 8: 807-21.
- 24. Ferrante RJ, Andreassen OA, Dedeoglu A, et al. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington’s disease. J Neurosci 2002; 22: 1592-9.
- 25. Rosas HD, Koroshetz WJ, Jenkins BG, et al. Riluzole therapy in Huntington’s disease. Mov Disord 1999; 14: 326-30.
- 26. Tabrizi SJ, Cleeter MW, Xuereb J, et al. Biochemical abnormalities and excitotoxicity in Huntington’s disease brain. Ann Neurol 1999; 45: 25-32.
- 27. Laforet GA, Sapp E, Chase K, et al. Changes in cortical and striatal neurons predict behavioral and electrophysiological abnormalities in a transgenic murine model of Huntington’s disease. J Neurosci 2001; 21: 9112-23.
- 28. Schilling G, Becher MW, Sharp AH, et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet 1999; 8: 397-407.
- 29. Shelbourne PF, Killeen N, Hevner RF, et al. A Huntington’s disease CAG expansion at the murine Hdh locus is unstable and associated with behavioural abnormalities in mice. Hum Mol Genet 1999; 8: 763-74.
- 30. Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 2000; 101: 57-66.
List of figures
Figure 1
Inclusions intranucléaires et agrégats extranucléaires dans le striatum d’une souris transgénique R6/2.
Ce marquage a été obtenu en utilisant un anticorps reconnaissant la partie amino-terminale de la huntingtine mutée. Les noyaux cellulaires sont colorés au vert de méthyle. La flèche indique une inclusion intranucléaire, et la pointe de flèche un agrégat extranucléaire. Ces deux types d’agrégats sont également présents dans le cerveau de patients atteints de chorée de Huntington. sb: substance blanche correspondant à la capsule interne traversant le striatum (photo, J.C. Liévens).
Figure 2
Mécanismes susceptibles de conduire au dysfonctionnement et à la mort des neurones dans la chorée de Huntington.
La présence d’une expansion de polyglutamine dans la huntingtine mutée entraîne son accumulation dans le noyau des neurones et la formation d’inclusions intranucléaires. Des protéines chaperons et des constituants du protéasome, d’une part, et des histone acétylases et des co-facteurs de transcription, d’autre part, seraient séquestrés dans ces inclusions ou interagiraient avec la partie amino-terminale de la huntingtine mutée. Cela aurait pour conséquence une accumulation de protéines mal conformées et un dérèglement de la transcription. De plus, la huntingtine mutée affecterait les fonctions mitochondriales. L’ensemble de ces altérations provoquerait un dysfonctionnement des cellules nerveuses. Selon une hypothèse, pour certains types de neurones comme les neurones efférents du striatum, un tel dysfonctionnement, affectant à la fois ces cellules, les neurones pré-synaptiques et les cellules gliales avoisinantes, entraînerait une augmentation des concentrations cytosoliques de calcium et de radicaux libres, notamment au travers de processus excitotoxiques. Ces effets pourraient être à l’origine de la mort neuronale.
List of tables
Tableau I
Modèles de souris transgéniques utilisés pour l’analyse de la chorée de Huntington.
Tableau I (continuation)
IT15: gène humain codant pour la huntingtine. CMV: cytomégalovirus. Les résultats obtenus avec ces modèles murins suggèrent que les déficits comportementaux observés dans la chorée de Huntington précèdent l’apparition des dégénérescences neuronales et sont probablement causés par un dysfonctionnement cellulaire. Aucune corrélation claire n’existe entre la mort neuronale et la mortalité des animaux, ni avec l’apparition des inclusions [2-8, 20, 27-30].