Article body
L’initiation de la traduction est une étape clé de la régulation de la synthèse protéique. Elle fait intervenir de nombreux facteurs dont l’organisation en complexes spécifiques permet la reconnaissance de l’ARNm par le ribosome [1]. Parmi ces facteurs, eIF2B (eukaryotic initiation factor 2B), constitué de 5 sous-unités protéiques désignées par les lettres α à ε et codées par les gènes EIF2B1 à B5 respectivement, a pour rôle d’activer le facteur eIF2 élément constitutif du complexe 43S de pré-initiation de la traduction. En effet, grâce à son activité d’échange GEF (guanine exchange factor) portée par la sous-unité ε, eIF2B contrôle l’activité du facteur eIF2, le faisant passer d’un état actif (lié au GTP) à inactif (lié au GDP) (Figure 1).
Figure 1
Activation du facteur eIF2 par le facteur eIF2B lors de l’étape d’initiation de la synthèse protéique.
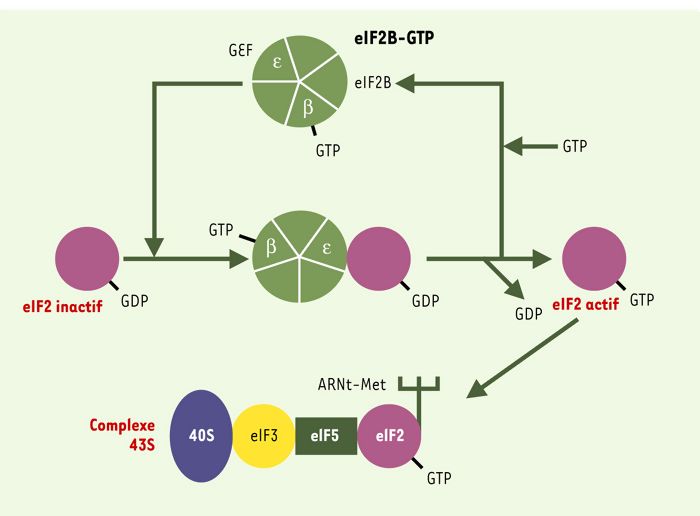
Le facteur d’initiation de la traduction eIF2 (violet) est activé par échange du GDP en GTP grâce au facteur activé eIF2B-GTP (en vert, composé de cinq sous-unités différentes). La sous-unité ε de eIF2B permet l’échange proprement dit du GDP en GTP (activité GEF: guanine exchange factor), la sous-unité β se lie au GTP. La forme activée eIF2-GTP se lie au complexe initiateur ARNt-méthionine, qui reconnaît spécifiquement le codon start AUG de l’ARNm, et est inclus avec d’autres facteurs d’initiation et la molécule 40S ribosomique dans le complexe protéique d’initiation 43S.
Ce facteur eIF2B a récemment été mis en cause dans différentes maladies héréditaires à transmission autosomique récessive touchant la substance blanche cérébrale (leucodystrophies) [2-4]. En effet, des mutations touchant l’ensemble des 5 gènes codant pour les 5 sous-unités protéiques d’eIF2B ont été identifiées chez des patients atteints du syndrome CACH (childood ataxia with central hypomyelinisation) ou VWM (leukoencephalopathy with vanishing white matter) [2]. Le syndrome CACH est une leucodystrophie progressive, préalablement identifiée sur des critères cliniques et sur des images caractéristiques à l’IRM, sous forme de zones cavitaires au sein d’une substance blanche massivement et précocement pathologique (Figure 2) [5]. La forme classique débute dans l’enfance, après un développement initial normal, par des troubles de la marche (syndrome cérébellospastique). L’évolution est émaillée de poussées d’aggravation brutale déclenchées par un traumatisme crânien bénin ou par une infection virale banale. L’atteinte cognitive est tardive. Le décès survient en général 3 à 5 ans après le début de la maladie. Des formes débutant plus tardivement pendant l’adolescence ou à l’âge adulte, révélées par des troubles cognitifs ou de l’humeur, ont été décrites et le facteur eIF2B a été impliqué. Leur évolution est lente, et l’aspect cavitaire de la substance blanche à l’IRM souvent discret ou initialement absent [6].
Figure 2
IRM et coupe d’un cerveau d’un patient atteint de CACH/VWM.
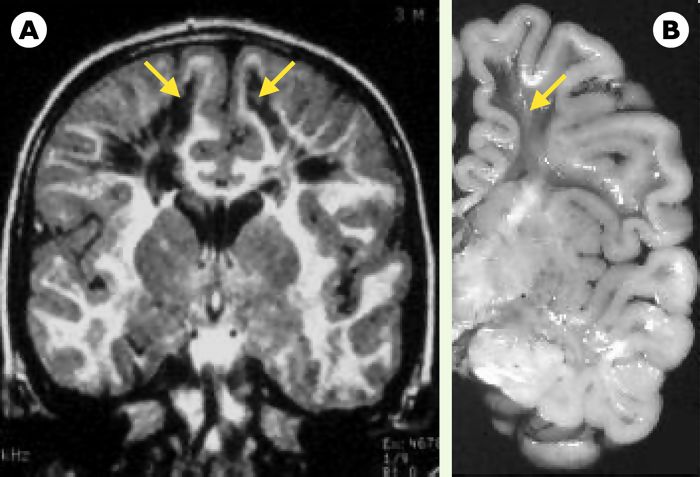
L’IRM (séquence FLAIR, A) comme la coupe anatomique du cerveau (B, cliché Dr A. Gelot) d’un patient atteint de CACH/VWM montrent de larges cavitations de la substance blanche (flèches jaunes).
Dans ces maladies, les mutations détectées dans les 5 gènes EIF2B sont majoritairement des mutations faux-sens. Dans notre expérience, la mutation R113H du gène EIF2B5, codant pour la sous-unité ε du facteur eIF2B, semble prédominante [2]. Aucune corrélation génotype-phénotype n’a été identifiée jusqu’à présent.
Plus récemment, nous avons pu mettre en évidence une forme à début très précoce (<1 an) avec une évolution rapide fatale en 6 mois [7], également liée à une mutation homozygote (Val309Leu) dans le gène EIF2B5 [4]. Nous avons pu rapprocher cette observation de celle d’une leucodystrophie décrite dans une population d’Indiens Cree du Mannitoba (CLE, Cree leukoencephalopathy) à début encore plus précoce (de 3 à 6 mois) entraînant le décès en quelques mois [8]. Nous avons identifié chez tous les malades une mutation homozygote dans le gène EIF2B5 (Arg195His), suggérant l’existence d’un effet fondateur dans cet isolat génétique [3]. Dans une étude ayant porté sur plus de 50 familles, cette mutation a également été identifiée à l’état hétérozygote dans une seule famille, dont certains membres étaient atteints de CACH/WVM, et qui était originaire du nord de l’Écosse. Or, dans les années 1700, le nord du Québec a connu une vague d’immigration européenne d’Écossais travaillant pour la Hudson Bay Company (voirEncadré), et des études ont montré que la maladie est apparue à cette époque dans la population Cree [9]. Notre étude moléculaire tend donc à conforter cette hypothèse.
Quel est le rôle du facteur eIF2B dans l’apparition d’une maladie limitée à la substance blanche cérébrale?
L’aggravation de la maladie que provoquent une infection virale fébrile ou un traumatisme crânien pourrait être expliquée par le rôle que joue le facteur eIF2B dans la réponse au stress cellulaire. En effet, lors d’un stress cellulaire, la traduction protéique doit être fortement réprimée afin d’éviter la synthèse de protéines dénaturées. Parallèlement à la production de protéines heat shock [10], l’induction de kinases spécifiques [11] permet la phosphorylation de la sous-unité α d’eIF2, ce qui accroît son affinité pour eIF2B et inhibe l’échange GDP/GTP: l’initiation de la synthèse protéique est ainsi diminuée. Les mutations du facteur eIF2B pourraient entraver ce mécanisme protecteur. L’accumulation de protéines dénaturées perpétuerait alors le stress imposé à la cellule [10].
Cela n’explique pas la sensibilité particulière de la substance blanche cérébrale à ces mutations. Des patients asymptomatiques ayant des anomalies de la substance blanche à l’IRM ont été décrits. Parmi les hypothèses possibles, on peut suggérer que les mutations du facteur eIF2B perturbent le processus de myélinisation lors du développement et/ou son maintien au niveau du système nerveux central par un mécanisme encore inconnu. Cette substance blanche « fragile » pourrait avoir une sensibilité accrue au stress cellulaire.
Ces résultats soulignent l’intérêt de la classification des leucodystrophies de cause indéterminée sur des critères cliniques et d’IRM, qui guiderait l’identification des gènes en cause. Reste à caractériser dans des études fonctionnelles le rôle exact d’eIF2B, ce qui pourrait déboucher sur des stratégies thérapeutiques permettant de prévenir ou de « stopper » l’évolution actuellement inéluctable de ces affections.

Appendices
Références
- 1. Ohlmann T, Derrington E, Lopez-Lastra M, et al. L’initiation de la synthèse des protéines chez les eucaryotes. Med Sci 2000; 16: 77-86.
- 2. Van Der Knaap MS, Leegwater PAJ, Könst AAM, et al. Mutations in each of the five subunits of translation initiation factor eIF2B can cause leukoencephalopathy with vanishing white matter. Ann Neurol 2002; 51: 264-70.
- 3. Fogli A, Wong K, Eymard-Pierre E, et al. Cree leukoencephalopathy and vanishing white matter disease are allelic at the eukaryotic translation initiation factor 2B5 locus. Ann Neurol 2002; 52: 506-10.
- 4. Fogli A, Dionisi-Vici C, Deodato F, et al. A severe variant of CACH/VWM leukoencephalopathy related to EIF2B5 mutation. Neurology, 2002; 59: 1966-8.
- 5. Van der Knaap MS, Barth PG, Gabreels FJ, et al. A new leukoencephalopathy with vanishing white matter. Neurology 1997; 48: 845-55.
- 6. Rodriguez D, Gelot A, Della GB, et al. Increased density of oligodendrocytes in childhood ataxia with diffuse central hypomyelination (CACH) syndrome: neuropathological and biochemical study of two cases. Acta Neuropathol 1999; 97: 469-80.
- 7. Francalanci P, Eymard-Pierre E, Dionisti-Vici C, et al. Fatal infantile leukodystrophy: a severe variant of CACH/VWM syndrome, allelic to chromosome 3q27. Neurology 2001; 57: 265-70.
- 8. Black DN, Booth F, Watters GV, et al. Leukoencephalopathy among native Indian infant in northern Quebec and Mannitoba. Ann Neurol 1988; 24: 490-6.
- 9. Francis D, Morantz T. Partners in Furs: a history of the Fur Trade in Eastern James Bay, 1600-1870. Montréal: McGill-Queen’s University Press, 1983.
- 10. Puolin F, Pyronnet S. Interactions moléculaires et initiation de la synthèse protéique. Med Sci 2000; 16: 617-22.
- 11. Webb BLJ, Proud CG. Eukaryotic initiation factor 2B (eIF2B). Int J Biochem Cell Biol 1997; 29: 1127-31.
List of figures
Figure 1
Activation du facteur eIF2 par le facteur eIF2B lors de l’étape d’initiation de la synthèse protéique.
Le facteur d’initiation de la traduction eIF2 (violet) est activé par échange du GDP en GTP grâce au facteur activé eIF2B-GTP (en vert, composé de cinq sous-unités différentes). La sous-unité ε de eIF2B permet l’échange proprement dit du GDP en GTP (activité GEF: guanine exchange factor), la sous-unité β se lie au GTP. La forme activée eIF2-GTP se lie au complexe initiateur ARNt-méthionine, qui reconnaît spécifiquement le codon start AUG de l’ARNm, et est inclus avec d’autres facteurs d’initiation et la molécule 40S ribosomique dans le complexe protéique d’initiation 43S.
Figure 2
IRM et coupe d’un cerveau d’un patient atteint de CACH/VWM.
L’IRM (séquence FLAIR, A) comme la coupe anatomique du cerveau (B, cliché Dr A. Gelot) d’un patient atteint de CACH/VWM montrent de larges cavitations de la substance blanche (flèches jaunes).