Abstracts
Résumé
Plusieurs protéines bactériennes décrites ces dernières années ont pour propriété commune d’inhiber la prolifération des cellules en culture, sans toutefois provoquer de cytotoxicité immédiate. De telles observations suggèrent l’existence de mécanismes bactériens de contrôle du cycle cellulaire eucaryote qui contribueraient au pouvoir pathogène ou à l’adaptation des souches à leur hôte. Ce concept émergent est analysé de manière critique, en prenant comme modèle une famille de toxines dont le mode d’action sur le cycle cellulaire a été récemment particulièrement bien étudié: les cytolethal distending toxins (CDT). Par le biais d’une probable altération de l’ADN, de nature encore indéterminée, ces protéines activent un point de contrôle (checkpoint) du cycle cellulaire, aboutissant au blocage des cellules en phase G2. Il existe encore peu de données expérimentales permettant d’extrapoler in vivo l’activité antiproliférative de ces protéines bactériennes, que nous proposons d’appeler cyclostatines, et dont le mode d’action est probablement très diversifié.
Summary
Several bacterial proteins have been recently described that share the ability to inhibit the proliferation of cells in culture without causing early signs of cytotoxicity. Such observations suggest the existence of bacterial mechanisms of control of the eukaryotic cell cycle contributing to pathogenicity or adaptation to the host. This emerging concept of cellular microbiology is critically analyzed considering as a model the cytolethal distending toxins (CDT), a family of toxins whose mode of action on the cell cycle has been thoroughly studied over the last few years. CDTs activate a physiological G2 checkpoint in exposed cells, probably from an initial DNA alteration whose precise molecular nature has not yet been determined. Experimental data are lacking to extrapolate in vivo the antiproliferative effect of these bacterial proteins that we tentatively propose to call cyclostatins.
Article body
La microbiologie cellulaire se propose d’étudier la capacité des bactéries à détourner à leur profit certaines voies de signalisation spécifiques des cellules de mammifères. La dynamique du cytosquelette est l’une des fonctions cellulaires les plus étudiées dans cette discipline, notamment en raison de son rôle essentiel dans les processus d’invasion et de transcytose bactérienne. De même, la modulation de la synthèse des cytokines constitue un autre champ majeur d’investigation, du fait de son impact sur les réponses immunitaire et inflammatoire de l’hôte.
Enfin, un nouveau domaine d’intérêt est sur le point d’émerger: celui de la manipulation du cycle cellulaire de l’hôte par les bactéries pathogènes. De nombreuses protéines bactériennes capables de bloquer ou de retarder spécifiquement la prolifération cellulaire ont en effet été décrites ces dernières années. L’existence de ces protéines soulève l’hypothèse, encore peu étayée in vivo, selon laquelle le ciblage des mécanismes de contrôle du cycle cellulaire constitue une stratégie de virulence ou d’adaptation des bactéries à l’hôte et leur confère un avantage sélectif. Nous proposons d’appeler cyclostatines les protéines bactériennes capables de bloquer ou de ralentir la prolifération cellulaire en agissant sur des voies de régulation du cycle.
Parmi les cyclostatines bactériennes possibles figure la famille des toxines CDT (cytolethal distending toxin), dont le mode d’action sur le cycle a été très étudié ces dernières années. Les toxines de cette famille, représentée dans plusieurs espèces bactériennes pathogènes, activent très efficacement une voie de signalisation cellulaire bloquant irréversiblement les cellules en phase G2 du cycle cellulaire. Cette famille de CDT peut être considérée comme un modèle de base, auquel on peut comparer d’autres protéines bactériennes inhibant la prolifération cellulaire selon des modalités variées, et dont les mécanismes sous-jacents sont souvent spéculatifs. Au préalable, quelques données générales sur la régulation du cycle cellulaire sont présentées, en relation avec leur exploitation possible par les bactéries. Les mécanismes de contrôle du cycle font l’objet de nombreuses synthèses récentes, notamment en langue française, auxquelles le lecteur est invité à se référer ((→) m/s 2003, n°2, p. 173; n° 3, p. 299 et p.309)[1, 2].
Voies de régulation du cycle cellulaire exploitables par les bactéries
Les mécanismes de régulation du cycle ont deux finalités principales: assurer l’intégrité du génome et sa parfaite reproduction dans les cellules filles après la division, et coupler la prolifération cellulaire aux contraintes de l’environnement externe. Cette régulation extrêmement complexe met en jeu des voies de signalisation interdépendantes dont on peut dégager différentes constantes.
En premier lieu, les effecteurs situés le plus en aval des cascades de régulation du cycle cellulaire sont les kinases dépendantes des cyclines (CDK). Les CDK contrôlent en particulier les stades critiques que constituent les transitions successives entre les phases G1, S, G2 et M du cycle cellulaire (Figure 1). Physiologiquement, l’activité des CDK au cours du cycle est corrélée à leur association avec leur partenaire régulatrice cycline, à leur niveau d’activation catalytique et à l’association du complexe CDK/cycline aux inhibiteurs de kinase cycline-dépendants (CKI) (Figure 2).
Figure 1
Complexes kinases dépendantes des cyclines (CDK)/cyclines et points de contrôle (PC).
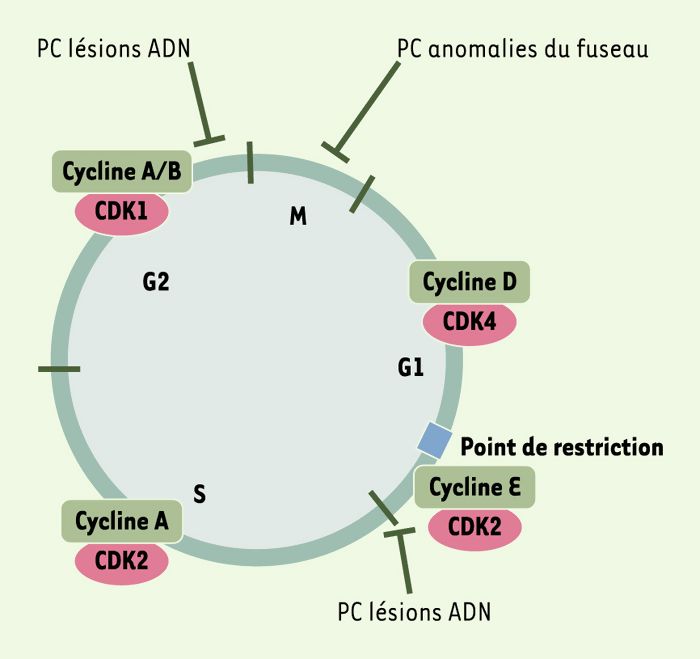
Les complexes CDK/cyclines réglent la progression des cellules dans le cycle [44]. Les cyclines mitotiques A et B s’accumulent pendant l’interphase, atteignant leur pic en mitose où elles sont dégradées par des mécanismes dépendant du protéasome. D’autres cyclines s’accumulent et culminent sur d’autres fenêtres du cycle, comme les cyclines D qui sont exprimées en G1 et dégradées lors de l’entrée en S. Les complexes CDK4/cycline D et CDK2/cycline E phosphorylent des substrats tels que la protéine pRB (protéine de susceptibilité au rétinoblastome), mettant ainsi en place les conditions requises pour la transition vers S. Le complexe CDK2/cycline A est nécessaire au contrôle de la phase S, tandis que l’activité des complexes CDK1/cycline A et CDK1/cycline B est essentielle pour déclencher les événements architecturaux et biochimiques qui caractérisent la mitose. Les points de contrôle peuvent être définis comme des voies biochimiques conditionnant la progression du cycle en un point particulier à la réussite d’une étape antérieure. Deux points de contrôle des lésions ADN sont présents en G1 et en G2, bloquant la progression du cycle en cas de lésion du génome ou de défaut dans le processus de réplication (voir aussi Figure 3 pour plus de détails). Par ailleurs, un point de contrôle détecte des anomalies du fuseau mitotique (SAC) ou des défauts d’attachement des chromosomes, ce qui conduit à l’inhibition de l’activité d’un complexe dit de promotion de l’anaphase. Le point de restriction correspond au mécanisme de contrôle qui permet aux cellules en G0/G1 d’entrer ou non dans le cycle en fonction de la concentration en facteurs de croissance.
Figure 2
Activation directe ou indirecte d’un point de contrôle du cycle cellulaire.
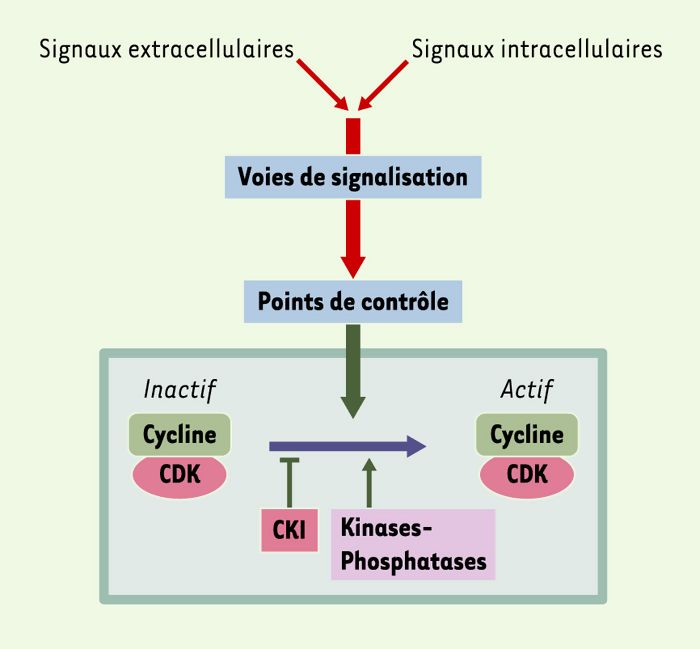
Ce schéma récapitule les voies de signalisation concourant, directement ou indirectement, à l’activation d’un point de contrôle, et pouvant théoriquement faire l’objet d’un détournement par une cyclostatine bactérienne. Chaque point de contrôle est une cascade complexe impliquant en amont des circuits de surveillance et de détection mis en oeuvre soit à chaque cycle, soit en cas de défaut accidentel, l’étape finale étant dans tous les cas l’activation d’un complexe CDK/cycline [45]. Des signaux extracellulaires (stress, facteurs de croissance, activation de récepteurs, attachement sur la matrice extracellulaire…) peuvent activer des cascades non liées directement au contrôle du cycle (voies des MAPK, des PKC ou des céramides), qui peuvent ensuite se connecter au point de contrôle à des étapes variées. L’étape finale d’activation des CDK est représentée avec ses acteurs principaux, c’est-à-dire, outre le complexe CDK/cycline lui-même, l’activateur de type kinase-phosphatase (telle que la phosphatase CDC25 qui agit principalement sur CDK1) et les inhibiteurs de CDK (ou CKI) (tels que la kinase WEE1 qui inhibe CDK1 en la phosphorylant [44]).
En second lieu, les cascades de régulation du cycle, même si leur déclenchement intervient plus en amont, aboutissent en général à un arrêt entre deux phases successives du cycle. On désigne sous le terme général de «points de contrôle» les voies biochimiques qui, à un stade donné, lient la progression du cycle à la réalisation complète d’une étape antérieure. Deux points de contrôle sont importants (Figure 1): celui des lésions de l’ADN, mécanisme détectant une altération de l’ADN, produisant un signal qui arrête les cellules (à la transition G1/S ou à la transition G2/M) et induisant la mise en place des mécanismes de réparation, et celui des anomalies du fuseau mitotique, qui détecte les défauts dans l’attachement des chromosomes au fuseau mitotique et bloque les cellules à la transition entre métaphase et anaphase. On peut également noter l’existence d’un point de restriction, mécanisme de contrôle requérant des CDK et des cyclines. Situé en fin de phase G1, le point de restriction correspond à la transition où la cellule s’autorise ou non à entrer dans un cycle de division (donc en premier lieu en S) en fonction de la présence de facteurs de croissance dans le milieu.
Enfin, dans certaines situations, d’autres médiateurs ou voies de signalisation peuvent se connecter aux points de contrôle spécialisés dans la régulation du cycle. C’est en particulier le cas de voies de signalisation relayant des signaux extracellulaires, telles que les voies des MAPK (mitogen-activated protein kinases), et en particulier des ERK (extracellular signal-regulated kinases) [3], des PKC (protéine kinase C) [4] et des céramides [5]. La progression du cycle peut ainsi être tributaire de facteurs extracellulaires tels que des stress ou des facteurs de croissance. Des signaux intracellulaires peuvent également interférer avec les voies de contrôle du cycle, comme le montre l’implication de la MAP kinase p38 dans le renforcement du point de contrôle des lésions de l’ADN en G2 [6] et du point de contrôle des anomalies du fuseau [7].
De cet ensemble de caractères généraux, il ressort qu’un grand nombre de cibles cellulaires sont théoriquement offertes aux bactéries pathogènes pour bloquer la prolifération cellulaire. Ces cibles potentielles peuvent être de nature membranaire, cytoplasmique ou nucléaire, et le «piratage» peut concerner tout ou partie d’un point de contrôle, ou encore affecter une voie secondaire connectée à ce point de contrôle (Figure 2). La microbiologie cellulaire ayant déjà mis en relief la très grande diversité des stratégies employées par les bactéries pour détourner les voies de régulation du cytosquelette, il n’est pas irréaliste de penser qu’il en pourrait en aller de même pour la régulation du cycle cellulaire.
CDT comme prototype des cyclostatines
Effet des toxines CDT sur le cycle cellulaire eucaryote
Les toxines CDT sont produites par plusieurs espèces bactériennes pathogènes Gram négatives: Escherichia coli, Campylobacter sp., Shigella dysenteriae, Actinobacillus actinomycetemcomitans, Haemophilus ducreyi, Helicobacter sp. [8]. Elles exercent sur les cellules épithéliales en culture un effet caractérisé par l’absence de figures de mitose et un grossissement important des cellules, la mortalité ne commençant significativement que 48 à 72 heures après l’exposition. Les cellules s’accumulent au stade G2 du cycle cellulaire et ne sont pas ralenties pendant leur progression en phase S. Ce blocage en G2 évoquant celui causé par certains agents génotoxiques, l’implication d’effecteurs clés de la cascade de signalisation dite du «point de contrôle en G2» (point de contrôle des lésions de l’ADN) (Figure 3), a été examinée.
Figure 3
Fonctionnement des point de contrôle des lésions de l’ADN.
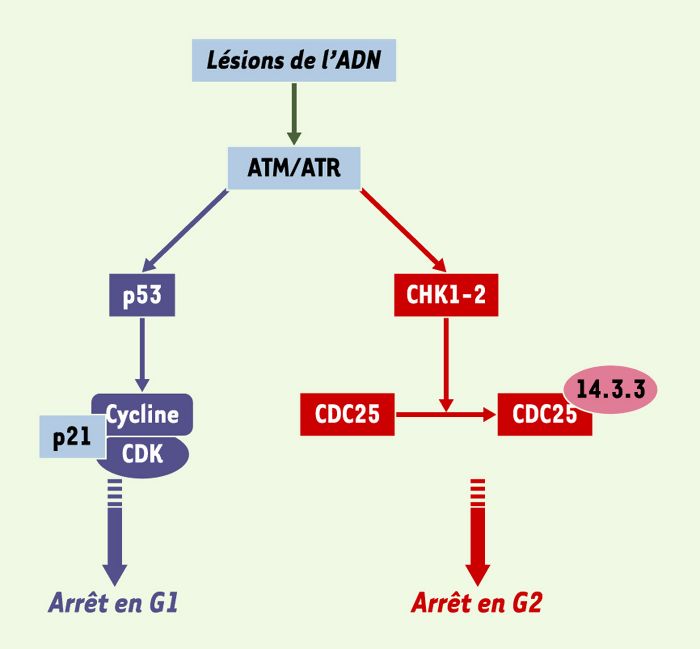
Le repérage des lésions de l’ADN par des complexes protéiques de détection (senseurs) conduit à l’activation des kinases ATM et ATR. Une première conséquence de cette activation est la phosphorylation de p53 (à gauche, en bleu), permettant l’accumulation de p21CiP1, inhibiteur des complexes CDK/cyclines à la transition G1/S [46]. L’autre conséquence possible (à droite, en rouge) est l’activation des kinases CHK1 et CHK2 qui phosphorylent la phosphatase CDC25. Cette modification, en permettant l’association de CDC25 avec des protéines de la famille 14.3.3, la retient dans le cytoplasme et l’empêche d’agir vis-à-vis des CDK nucléaires. Les complexes CDK/cycline sont donc inactifs, ce qui empêche les cellules de franchir la transition G2/M.
En premier lieu, l’implication du complexe formé par la kinase dépendante des cyclines CDK1 et par son partenaire régulateur, la cycline B, a été étudiée. Ce complexe contrôle en effet directement la transition entre le stade G2 et la mitose [9] (Figure 1). Il est présent à une concentration normale dans les cellules bloquées par une toxine CDT, mais CDK1 est sous sa forme inactive hyperphosphorylée, ce qui rend parfaitement compte de l’inhibition du déclenchement de la mitose. CDT n’agit toutefois pas comme inhibiteur direct de CDK1, puisqu’on peut restaurer la forme active de CDK1 par action de la phosphatase CDC25 (in vitro comme dans les cellules elles-mêmes) [10, 11]. Les phosphatases CDC25, effecteurs situés directement en amont de CDK1, sont responsables de la déphosphorylation, et donc de l’activation de ces kinases CDK. La possibilité que CDT active des effecteurs situés en amont de CDK1 dans la cascade a donc été recherchée. En étudiant le comportement, vis-à-vis de la toxine, des cellules mutées dans les gènes correspondants [12, 13], on a pu mettre en évidence que CDT provoque l’activation par phosphorylation de la kinase CHK2 (checkpoint kinase2), puis fortement suspecter qu’elle entraîne celle de la kinase ATM.
On peut conclure de ces résultats que CDT active un point de contrôle en G2 du cycle cellulaire présentant des analogies frappantes avec la cascade dite des dommages de l’ADN. Cette hypothèse est renforcée par le fait que l’exposition des cellules à CDT entraîne, comme avec les radiations ionisantes, un recrutement nucléaire précoce des protéines H2AX et Mre11 du complexe dit de réparation de l’ADN [14]. Bien que la réalité des dommages de l’ADN n’ait pas été démontrée avec des concentrations de toxine capables de bloquer le cycle en G2, ces observations soutiennent l’hypothèse que les effets de CDT sur le cycle cellulaire eucaryote sont bien liés à une modification de l’ADN.
Relation structure-fonction dans les protéines trimériques CDT
Les CDT forme une famille hétérogène de toxines présentes dans un large spectre d’espèces bactériennes Gram négatives pathogènes chez l’homme et les animaux [8]. Toutes les toxines CDT sont codées par un locus de trois gènes (cdt A, B et C) organisés en un seul opéron, et codant pour des protéines de taille comprise entre 15 et 30 kDa. Le degré d’identité entre gènes de toxines d’origine différente est très variable, le gène cdtB étant de loin le plus conservé. La présence simultanée des trois gènes est indispensable à l’expression de CDT [15-18], de même qu’il n’est possible de reconstituer l’activité de CDT qu’à condition de mélanger les trois protéines clonées séparément [19-22]. Cela signifie que chaque sous-unité joue un rôle indispensable dans l’une ou l’autre des étapes de l’interaction de la toxine avec la cellule-cible: reconnaissance d’un récepteur périphérique, internalisation et trafic intracellulaire, translocation nucléaire et enfin activité catalytique sur la cible nucléaire. Aucune de ces étapes n’a cependant été directement mise en évidence à ce jour. La seule certitude est que cdt B code pour la sous-unité catalytique de la toxine. Cette conclusion, corroborée par le fait que CDT-B est la protéine la plus conservée, est fondée sur les analogies lointaines que la molécule présente avec des enzymes de la grande famille des phosphodiestérases [8], et sur la démonstration que la mutation d’acides aminés critiques des sites enzymatiques présumés abroge l’activité de la toxine [23, 24]. L’hypothèse la plus plausible est que CDT-B est une nucléase, comme certains membres de la famille des phosphodiestérases tels que la DNAse-I. En effet, CDT-B exerce in vitro, sur l’ADN, et à très forte dose, une activité de coupure de brin se traduisant par le relargage de plasmides superenroulés [25]. De même, l’expression forcée de CDT-B à l’intérieur de cellules après transfection ou micro-injection conduit à la translocation nucléaire de la molécule, puis à des signes de dégradation de la chromatine [23, 26].
En résumé, les observations rapportées à ce jour sur les voies de signalisation activées par CDT comme sur les propriétés de la sous-unité catalytique CDT-B permettent de dresser un tableau cohérent du mode d’action de CDT sur les cellules eucaryotes (Figure 4): CDT-B modifierait l’ADN nucléaire, ce qui déclencherait l’activation du complexe de réparation auquel sont associées les protéines H2AX et Mre11, ainsi qu’une cascade de contrôle du cycle en G2 passant successivement par ATM, CHK2, CDC25 et enfin CDK1. Les principales inconnues restant à élucider sont la nature exacte des modifications infligées à l’ADN et le rôle respectif des sous-unités CDT-A et CDT-C, probablement dédiées aux phases d’attachement spécifique et d’internalisation de la toxine.
Figure 4
Contrôle du cycle cellulaire par CDT.
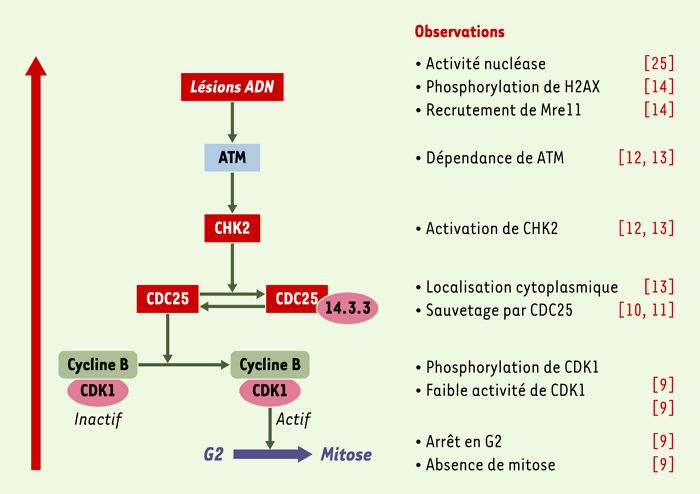
Récapitulatif des étapes de mise en évidence de l’implication des effecteurs du point de contrôle en G2 des lésions de l’ADN dans la signalisation induite par CDT dans les cellules de mammifères.
Rôle de CDT dans le pouvoir pathogène
Les CDT sont produites par des espèces bactériennes pathogènes des muqueuses (intestinale pour E. coli, Shigella, Campylobacter et Helicobacter; génitale pour H. ducreyi; gingivale pour A. actinomycetemcomitans). À l’heure actuelle, la contribution exacte de CDT au pouvoir pathogène des bactéries est encore largement spéculative, de même que la pertinence in vivo de son effet sur la prolifération cellulaire. L’existence de CDT dans plusieurs espèces bactériennes non apparentées, observation originale pour une toxine bactérienne, laisse penser que CDT confère un fort avantage sélectif aux bactéries qui la produisent. Que cet avantage soit lié à l’aptitude de CDT à contrôler la multiplication cellulaire des tissus cibles est très probable, bien qu’aucune démonstration ne soit encore venue étayer cette hypothèse. On ne peut pas retenir l’éventuel effet génotoxique de CDT comme un mécanisme de pathogénie en soi, car il serait très modéré et sans conséquence cytotoxique directe, ainsi que le suggèrent les résultats obtenus en cultures cellulaires dans des conditions physiologiques d’exposition à la toxine [10]. En revanche, il est plausible que les cibles majeures de CDT soient les tissus où la réponse proliférative est essentielle, comme les muqueuses et les épithéliums, dont le taux de renouvellement est très élevé et au sein desquels les (nombreux) lymphocytes doivent être mobilisables en permanence comme première barrière immunologique. De ce point de vue, les muqueuses digestive, gingivale et génitale, que colonisent préférentiellement les bactéries productrices de CDT, sont donc des tissus potentiellement sensibles car ils associent ces deux types cellulaires.
En dehors des effets des CDT in vivo sur la prolifération cellulaire, qui ne sont pas documentés, il existe des résultats expérimentaux obtenus sur cellules primaires et dans des modèles animaux, destinés à évaluer le rôle de CDT dans le pouvoir pathogène. En ce qui concerne les cellules primaires, CDT inhibe efficacement la réponse proliférative des lymphocytes B et T activés, notamment humains, en les bloquant en phase G2. Ces observations ont été effectuées avec des CDT d’origine différente, provenant notamment de A. actinomycetemcomitans et H. ducreyi, deux agents ayant en commun de provoquer des infections localisées, respectivement des muqueuses gingivale et vaginale, dont l’aggravation est liée au débordement de la réponse immunitaire locale.
En ce qui concerne les modèles animaux, l’inoculation par voie orale de la toxine CDT de S. dysenteriae provoque une diarrhée chez les souriceaux nouveau-nés [27], et CDT contribue aux propriétés invasives de C. jejuni chez la souris après colonisation digestive [28]. Dans ces deux études, aucune relation n’a été recherchée entre le phénomène de diarrhée ou d’invasion et l’état de prolifération (ou de différenciation) des entérocytes. Enfin, le rôle de CDT dans la formation de l’ulcère chancroïde génital provoqué par H. ducreyi a été étudié dans deux modèles cutanés (chez l’homme et chez le lapin) qui reproduisent la pustule, premier stade de cette lésion [19, 29, 30]. Les résultats montrent que CDT n’a aucun rôle déterminant dans la formation de la pustule, sa participation restant possible dans la formation de l’ulcère terminal.
Autres protéines bactériennes inhibant la prolifération cellulaire
Comme nous venons de le voir, la toxine CDT active le point de contrôle en G2 des lésions de l’ADN, de son origine, probablement une modification de l’ADN, jusqu’à son effecteur final, la kinase dépendante des cyclines CDK1. D’autres protéines bactériennes ayant en commun d’inhiber la prolifération des cellules en culture sans exercer d’effet cytotoxique précoce dans les mêmes cellules ont été décrites (Tableau I). Pour la plupart d’entre elles, la signalisation expliquant l’inhibition de prolifération n’est pas connue, notamment en ce qui concerne l’implication des CDK, effecteurs clés en aval des point de contrôles. Il est donc encore prématuré de faire une typologie fonctionnelle de ces produits.
Tableau I
Exemples de protéines bactériennes inhibant la prolifération cellulaire.
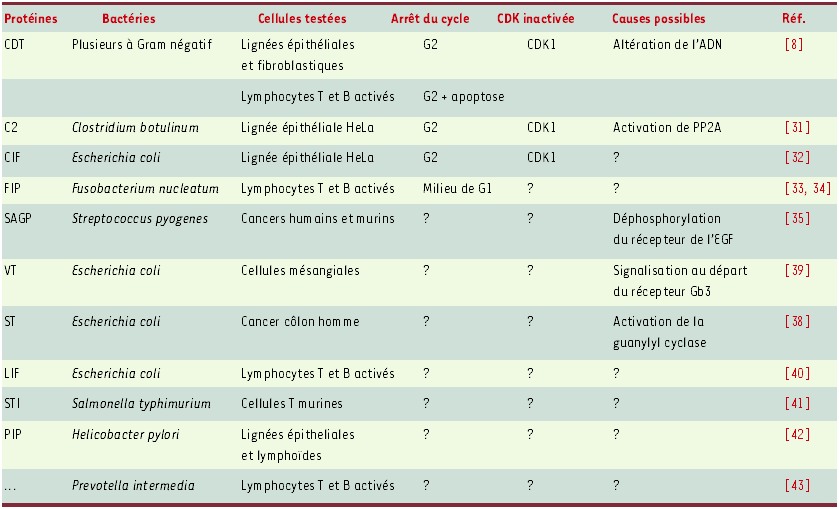
Facteurs bactériens : CDT : cytolethal distending toxin ; VT : verotoxin ; ST : thermostable enterotoxin ; SAGP : streptococcal antitumor glycoprotein ; FIP : fusobacterium immunosuppressive protein ; CIF : cycle inhibiting factor ; LIF : lymphocyte inhibiting factor (ou lymphostatine) ; STI : Salmonella typhimurium-derived T-cell inhibitor ; PIP : proliferation inhibiting protein ; PP2A : protein phosphatase 2A ; EGF : epidermal growth factor ; Gb3 : globotriaosylcéramide.
Deux de ces protéines, à l’instar de CDT, provoquent dans les cellules de type épithélial un blocage en G2 associé à une inactivation de CDK1 par hyperphosphorylation. Il s’agit de la toxine C2 de Clostridium botulinum [31] et de la protéine CIF d’E. coli [32]. Ces protéines semblent activer la dernière étape du point de contrôle des lésions de l’ADN, mais cela ne signifie pas pour autant qu’elles l’activent dans son intégralité, c’est-à-dire à partir d’une altération de l’ADN ou de perturbations dans la réplication. Pour le démontrer, il faudrait, comme pour CDT, vérifier l’implication des effecteurs en amont comme CDC25, les kinases CHK et ATM, ou encore démontrer la réalité des dommages de l’ADN dans les cellules exposées. Par ailleurs, il ne faut pas exclure la possibilité que ces blocages en G2 liés à une inactivation de CDK1 par phosphorylation soient induits par des voies alternatives connectées au point de contrôle. Ainsi l’hypothèse a-t-elle été émise selon laquelle la toxine C2, connue pour activer in vitro les tyrosine phosphatases, pourrait activer directement dans les cellules la protéine phosphatase 2A (PP2A), elle-même puissante inactivatrice de la phosphatase CDC25. Ce faisant, elle empêcherait l’activation de CDK1 par tyrosine déphosphorylation, inhibant ainsi le déclenchement de la mitose.
La protéine FIP de Fusobacterium nucleatum bloque en G1 les lymphocytes activés par des mitogènes ou des antigènes [33, 34]. Une étude de l’expression des cyclines spécifiques de cette phase montre que le stade d’arrêt ne coïncide pas avec le point de restriction, mais se situe entre celui-ci et l’entrée en phase S. Pour déterminer la cause exacte de ce blocage en G1, il faudrait commencer par examiner l’activité kinase des complexes CDK/cycline ayant un rôle spécifique au stade G1, notamment pour préparer la transition G1/S, à savoir les complexes CDK4/cyclines D et CDK2/cycline E. En cas d’inhibition de l’un de ces complexes, il faudrait ensuite examiner l’existence d’une interaction avec des CKI propres à ce stade, comme p16lnk4A ou p27kip1.
Pour les autres protéines figurant dans le Tableau I, le stade du cycle où les cellules sont bloquées n’est pas rapporté par les auteurs, mais des informations spécifiques sur les cibles cellulaires ou les voies de transduction activées sont indiquées. Ainsi, la glycoprotéine SAGP de Streptococcus pyogenes produit un arrêt de prolifération associé à une déphosphorylation du récepteur au facteur de croissance épidermique (EGF), puis une inhibition de la MAPK p42/44 [35]. Il resterait à élucider comment, dans ce cas particulier, la voie des MAPK se connecte à un point de contrôle (en G1 ou G2), par comparaison avec les blocages en G2 liés à l’EGF ou d’autres facteurs de croissance [36]. SAGP est la seule protéine présentée dans le Tableau I dont l’action anti-proliférative ait été démontrée in vivo, en l’occurrence vis-à-vis de tumeurs syngéniques de la souris [37]. Les propriétés pathogènes des deux toxines colibacillaires ST (entérotoxine thermostable) et VT (vérotoxine) sont bien connues. Plus originale est l’observation récente que ces deux toxines ralentissent fortement la prolifération de cellules constituant des cibles réelles in vivo, à savoir une lignée entérocytaire différenciée, pour ST [38], et des cellules mésangiales de glomérules rénaux, pour VT [39]. ST agissant comme un agoniste de la guanylyl cyclase, il est possible que son effet antiprolifératif sur les entérocytes soit dépendant d’une voie de transduction à laquelle participe la GMP-cyclique. Quant à VT, l’hypothèse avancée est que la signalisation conduisant à l’inhibition de prolifération pourrait démarrer du récepteur membranaire de la toxine, le glycosphingolipide Gb3.
Enfin un dernier groupe de protéines ont été caractérisées uniquement en référence à leur effet d’inhibition de la prolifération des lymphocytes activés, sans précision sur le stade du cycle où s’effectue le blocage: le LIF d’E. coli [40], le STI de Salmonella typhimurium [41], le PIP d’Helicobacter pylori [42] et une protéine de Prevotella intermedia [43].
Conclusions: vers une définition des cyclostatines bactériennes
À partir des informations rapportées dans cet article, on peut provisoirement définir les cyclostatines comme étant des protéines bactériennes provoquant un arrêt de prolifération cellulaire en interférant spécifiquement avec des effecteurs de voies de contrôle du cycle, en particulier les CDK. Certaines des protéines présentées sur le Tableau I ne répondent pas encore à cette définition, car la description de leur effet sur le cycle et sur ses effecteurs est encore trop sommaire.
Il faudrait ajouter à ces critères la démonstration d’un effet antiprolifératif in vivo. Or, contrairement à la plupart des concepts de la microbiologie cellulaire, tel que celui d’internalisation des bactéries dans les cellules, celui de cyclostatines est né d’observations faites à partir de cultures cellulaires et non pas d’animaux infectés. Il est donc essentiel d’en vérifier à présent la pertinence in vivo, en d’autres termes de démontrer la réalité des phénomènes antiprolifératifs dans des tissus cibles et d’en analyser les implications pour le pouvoir pathogène ou l’adaptation à l’hôte, ceci pour chaque espèce bactérienne productrice. La nécessité de trouver une finalité biologique aux nombreuses cyclostatines potentielles décrites dans la littérature est d’autant plus importante que les mécanismes conduisant à l’arrêt du cycle semblent à première vue très diversifiés. Cette exigence concerne en particulier la cyclostatine-type CDT dont la diffusion remarquable dans plusieurs espèces bactériennes pathogènes non apparentées est un argument fort en faveur d’un rôle dans le pouvoir pathogène ou dans l’adaptation aux hôtes.


Appendices
Références
- 1. Viallard JF, Lacombe F, Belloc F, Pellegrin JL, Reiffers J. Mécanismes moléculaires contrôlant le cycle cellulaire: aspects fondamentaux et implications en oncologogie. Cancer Radiother 2001; 5: 109-29.
- 2. Thuret JY, Mann C. Kinases activatrices de la division cellulaire: in vivo veritas ? Med Sci 1998; 14: 215-8.
- 3. Hulleman E, Boonstra J. Regulation of G1 phase progression by growth factors and the extracellular matrix. Cell Mol Life Sci 2001; 58: 80-93.
- 4. Fishman DD, Segal S, Livneh E. The role of protein kinase C in G1 and G2/M phases of the cell cycle. Int J Oncol 1998; 12: 181-6.
- 5. Jayadev S, Liu B, Bielawska AE, et al. Role for ceramide in cell cycle arrest. J Biol Chem 1995; 270: 2047-52.
- 6. Pearce AK, Humphrey TC. Integrating stress-response and cell-cycle checkpoint pathways. Trends Cell Biol 2001; 11: 426-33.
- 7. Takenaka K, Moriguchi T, Nishida E. Activation of the protein kinase p38 in the spindle assembly checkpoint and mitotic arrest. Science 1998; 280: 599-602.
- 8. De Rycke J, Oswald E. Cytolethal distending toxin (CDT): a bacterial weapon to control host cell proliferation? FEMS Microbiol Lett 2001; 203: 141-8.
- 9. Comayras C, Tasca C, Peres SY, Ducommun B, Oswald E, De Rycke J. Escherichia coli cytolethal distending toxin blocks the HeLa cell cycle at the G2/M transition by preventing cdc2 protein kinase dephosphorylation and activation. Infect Immun 1997; 65: 5088-95.
- 10. Sert V, Cans C, Tasca C, et al. The bacterial cytolethal distending toxin (CDT) triggers a G2 cell cycle checkpoint in mammalian cells without preliminary induction of DNA strand breaks. Oncogene 1999; 18: 6296-304.
- 11. Escalas N, Davezac N, De Rycke J, Baldin V, Mazars R, Ducommun B. Study of the cytolethal distending toxin-induced cell cycle arrest in HeLa cells: involvement of the CDC25 phosphatase. Exp Cell Res 2000; 257: 206-12.
- 12. Cortes-Bratti X, Karlsson C, Lagergard T, Thelestam M, Frisan T. The Haemophilus ducreyi cytolethal distending toxin induces cell cycle arrest and apoptosis via the DNA damage checkpoint pathways. J Biol Chem 2001; 276: 5296-302.
- 13. Alby F, Mazars R, De Rycke J, et al. Study of the cytolethal distending toxin (CDT)-activated cell cycle checkpoint. Involvement of the CHK2 kinase. FEBS Lett 2001; 491: 261-5.
- 14. Li L, Sharipo A, Chaves-Olarte E, et al. The Haemophilus ducreyi cytolethal distending toxin activates sensors of DNA damage and repair complexes in proliferating and non-proliferating cells. Cell Microbiol 2002; 4: 87-99.
- 15. Sugai M, Kawamoto T, Peres SY, et al. The cell cycle-specific growth-inhibitory factor produced by Actinobacillus actinomycetemcomitans is a cytolethal distending toxin. Infect Immun 1998; 66: 5008-19.
- 16. Peres SY, Marches O, Daigle F, et al. A new cytolethal distending toxin (CDT) from Escherichia coli producing CNF2 blocks HeLa cell division in G2/M phase. Mol Microbiol 1997; 24: 1095-107.
- 17. Pickett CL, Cottle DL, Pesci EC, Bikah G. Cloning, sequencing, and expression of the Escherichia coli cytolethal distending toxin genes. Infect Immun 1994; 62: 1046-51.
- 18. Scott DA, Kaper JB. Cloning and sequencing of the genes encoding Escherichia coli cytolethal distending toxin. Infect Immun 1994; 62: 244-51.
- 19. Lewis DA, Stevens MK, Latimer JL, et al. Characterization of Haemophilus ducreyi cdtA, cdtB, and cdtC mutants in in vitro and in vivo systems. Infect Immun 2001; 69: 5626-34.
- 20. Lara-Tejero M, Galan JE. CdtA, CdtB, and CdtC form a tripartite complex that is required for cytolethal distending toxin activity. Infect Immun 2001; 69: 4358-65.
- 21. Frisk A, Lebens M, Johansson C, et al. The role of different protein components from the Haemophilus ducreyi cytolethal distending toxin in the generation of cell toxicity. Microb Pathog 2001; 30: 313-24.
- 22. Deng K, Latimer JL, Lewis DA, Hansen EJ. Investigation of the interaction among the components of the cytolethal distending toxin of Haemophilus ducreyi. Biochem Biophys Res Commun 2001; 285: 609-15.
- 23. Lara-Tejero M, Galan JE. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science 2000; 290: 354-7.
- 24. Elwell CA, Dreyfus LA. DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol Microbiol 2000; 37: 952-63.
- 25. Elwell C, Chao K, Patel K, Dreyfus L. Escherichia coli CdtB mediates cytolethal distending toxin cell cycle arrest. Infect Immun 2001; 69: 3418-22.
- 26. Hassane DC, Lee RB, Mendenhall MD, Pickett CL. Cytolethal distending toxin demonstrates genotoxic activity in a yeast model. Infect Immun 2001; 69: 5752-9.
- 27. Okuda J, Fukumoto M, Takeda Y, Nishibuchi M. Examination of diarrheagenicity of cytolethal distending toxin: suckling mouse response to the products of the cdtABC genes of Shigella dysenteriae. Infect Immun 1997; 65: 428-33.
- 28. Purdy D, Buswell CM, Hodgson AE, McAlpine K, Henderson I, Leach SA. Characterisation of cytolethal distending toxin (CDT) mutants of Campylobacter jejuni. J Med Microbiol 2000; 49: 473-9.
- 29. Stevens MK, Latimer JL, Lumbley SR, et al. Characterization of a Haemophilus ducreyi mutant deficient in expression of cytolethal distending toxin. Infect Immun 1999; 67: 3900-8.
- 30. Young RS, Fortney KR, Gelfanova V, et al. Expression of cytolethal distending toxin and hemolysin is not required for pustule formation by Haemophilus ducreyi in human volunteers. Infect Immun 2001; 69: 1938-42.
- 31. Barth H, Klingler M, Aktories K, Kinzel V. Clostridium botulinum C2 toxin delays entry into mitosis and activation of p34cdc2 kinase and cdc25-C phosphatase in HeLa cells. Infect Immun 1999; 67: 5083-90.
- 32. Nougayrede JP, Marches O, Boury M, et al. The long-term cytoskeletal rearrangement induced by rabbit enteropathogenic Escherichia coli is Esp dependent but intimin independent. Mol Microbiol 1999; 31: 19-30.
- 33. Demuth DR, Savary R, Golub E, Shenker BJ. Identification and analysis of fipA, a Fusobacterium nucleatum immunosuppressive factor gene. Infect Immun 1996; 64: 1335-41.
- 34. Shenker BJ, Datar S. Fusobacterium nucleatum inhibits human T-cell activation by arresting cells in the mid-G1 phase of the cell cycle. Infect Immun 1995; 63: 4830-6.
- 35. Yoshida J, Ishibashi T, Nishio M. Growth-inhibitory effect of a streptococcal antitumor glycoprotein on human epidermoid carcinoma A431 cells: involvement of dephosphorylation of epidermal growth factor receptor. Cancer Res 2001; 61: 6151-7.
- 36. Gutkind JS. The pathways connecting G protein-coupled receptors to the nucleus through divergent mitogen-activated protein kinase cascades. J Biol Chem 1998; 273: 1839-42.
- 37. Yoshida J, Takamura S, Nishio M. Characterization of a streptococcal antitumor glycoprotein (SAGP). Life Sci 1998; 62: 1043-53.
- 38. Pitari GM, Di Guglielmo MD, Park J, Schulz S, Waldman SA. Guanylyl cyclase C agonists regulate progression through the cell cycle of human colon carcinoma cells. Proc Natl Acad Sci USA 2001; 98: 7846-51.
- 39. Van Setten PA, van Hinsbergh VW, Van den Heuvel LP, et al. Verocytotoxin inhibits mitogenesis and protein synthesis in purified human glomerular mesangial cells without affecting cell viability: evidence for two distinct mechanisms. J Am Soc Nephrol 1997; 8: 1877-88.
- 40. Klapproth JM, Scaletsky IC, McNamara BP, et al. A large toxin from pathogenic Escherichia coli strains that inhibits lymphocyte activation. Infect Immun 2000; 68: 2148-55.
- 41. Matsui K, Nagano K, Arai T, Hirono I, Aoki T. DNA sequencing of the gene encoding Salmonella typhimurium-derived T-cell inhibitor (STI) and characterization of the gene product, cloned STI. FEMS Immunol Med Microbiol 1998; 22: 341-9.
- 42. Knipp U, Birkholz S, Kaup W, Opferkuch W. Partial characterization of a cell proliferation-inhibiting protein produced by Helicobacter pylori. Infect Immun 1996; 64: 3491-6.
- 43. Shenker BJ, Vitale L, Slots J. Immunosuppressive effects of Prevotella intermedia on in vitro human lymphocyte activation. Infect Immun 1991; 59: 4583-9.
- 44. Grana X, Reddy EP. Cell cycle control in mammalian cells: role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene 1995; 11: 211-9.
- 45. Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science 1996; 274: 1664-72.
- 46. Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature 2000; 408: 433-9.
List of figures
Figure 1
Complexes kinases dépendantes des cyclines (CDK)/cyclines et points de contrôle (PC).
Les complexes CDK/cyclines réglent la progression des cellules dans le cycle [44]. Les cyclines mitotiques A et B s’accumulent pendant l’interphase, atteignant leur pic en mitose où elles sont dégradées par des mécanismes dépendant du protéasome. D’autres cyclines s’accumulent et culminent sur d’autres fenêtres du cycle, comme les cyclines D qui sont exprimées en G1 et dégradées lors de l’entrée en S. Les complexes CDK4/cycline D et CDK2/cycline E phosphorylent des substrats tels que la protéine pRB (protéine de susceptibilité au rétinoblastome), mettant ainsi en place les conditions requises pour la transition vers S. Le complexe CDK2/cycline A est nécessaire au contrôle de la phase S, tandis que l’activité des complexes CDK1/cycline A et CDK1/cycline B est essentielle pour déclencher les événements architecturaux et biochimiques qui caractérisent la mitose. Les points de contrôle peuvent être définis comme des voies biochimiques conditionnant la progression du cycle en un point particulier à la réussite d’une étape antérieure. Deux points de contrôle des lésions ADN sont présents en G1 et en G2, bloquant la progression du cycle en cas de lésion du génome ou de défaut dans le processus de réplication (voir aussi Figure 3 pour plus de détails). Par ailleurs, un point de contrôle détecte des anomalies du fuseau mitotique (SAC) ou des défauts d’attachement des chromosomes, ce qui conduit à l’inhibition de l’activité d’un complexe dit de promotion de l’anaphase. Le point de restriction correspond au mécanisme de contrôle qui permet aux cellules en G0/G1 d’entrer ou non dans le cycle en fonction de la concentration en facteurs de croissance.
Figure 2
Activation directe ou indirecte d’un point de contrôle du cycle cellulaire.
Ce schéma récapitule les voies de signalisation concourant, directement ou indirectement, à l’activation d’un point de contrôle, et pouvant théoriquement faire l’objet d’un détournement par une cyclostatine bactérienne. Chaque point de contrôle est une cascade complexe impliquant en amont des circuits de surveillance et de détection mis en oeuvre soit à chaque cycle, soit en cas de défaut accidentel, l’étape finale étant dans tous les cas l’activation d’un complexe CDK/cycline [45]. Des signaux extracellulaires (stress, facteurs de croissance, activation de récepteurs, attachement sur la matrice extracellulaire…) peuvent activer des cascades non liées directement au contrôle du cycle (voies des MAPK, des PKC ou des céramides), qui peuvent ensuite se connecter au point de contrôle à des étapes variées. L’étape finale d’activation des CDK est représentée avec ses acteurs principaux, c’est-à-dire, outre le complexe CDK/cycline lui-même, l’activateur de type kinase-phosphatase (telle que la phosphatase CDC25 qui agit principalement sur CDK1) et les inhibiteurs de CDK (ou CKI) (tels que la kinase WEE1 qui inhibe CDK1 en la phosphorylant [44]).
Figure 3
Fonctionnement des point de contrôle des lésions de l’ADN.
Le repérage des lésions de l’ADN par des complexes protéiques de détection (senseurs) conduit à l’activation des kinases ATM et ATR. Une première conséquence de cette activation est la phosphorylation de p53 (à gauche, en bleu), permettant l’accumulation de p21CiP1, inhibiteur des complexes CDK/cyclines à la transition G1/S [46]. L’autre conséquence possible (à droite, en rouge) est l’activation des kinases CHK1 et CHK2 qui phosphorylent la phosphatase CDC25. Cette modification, en permettant l’association de CDC25 avec des protéines de la famille 14.3.3, la retient dans le cytoplasme et l’empêche d’agir vis-à-vis des CDK nucléaires. Les complexes CDK/cycline sont donc inactifs, ce qui empêche les cellules de franchir la transition G2/M.
Figure 4
Contrôle du cycle cellulaire par CDT.
Récapitulatif des étapes de mise en évidence de l’implication des effecteurs du point de contrôle en G2 des lésions de l’ADN dans la signalisation induite par CDT dans les cellules de mammifères.
List of tables
Tableau I
Exemples de protéines bactériennes inhibant la prolifération cellulaire.
Facteurs bactériens : CDT : cytolethal distending toxin ; VT : verotoxin ; ST : thermostable enterotoxin ; SAGP : streptococcal antitumor glycoprotein ; FIP : fusobacterium immunosuppressive protein ; CIF : cycle inhibiting factor ; LIF : lymphocyte inhibiting factor (ou lymphostatine) ; STI : Salmonella typhimurium-derived T-cell inhibitor ; PIP : proliferation inhibiting protein ; PP2A : protein phosphatase 2A ; EGF : epidermal growth factor ; Gb3 : globotriaosylcéramide.