Abstracts
Résumé
Le ciblage de la voie d’endocytose constitue l’un des moyens développés par certains virus, le virus de l’immunodéficience humaine (VIH) et le virus de l’Herpès associé au sarcome de Kaposi (KSHV), pour moduler l’expression de surface de protéines notamment impliquées dans la réponse immunitaire. Cet article a pour but de faire le point sur les mécanismes que ces deux virus ont mis oeuvre, par l’intermédiaire des protéines Nef (VIH) et K3/K5 (KHSV), pour « parasiter » cette voie de transport intracellulaire. Les travaux récents montrent qu’effectivement leurs modes d’action sont bien plus complexes qu’initialement supposés.
Summary
The modulation of plasma membrane proteins involved in the communication with the immune system is a general mechanism developed by viruses to escape the immune response. Most of the studied examples have focused on viral proteins that missort cellular proteins during their biosynthesis. However, an increasing number of examples show that the down-modulation can also be achieved after membrane delivery by targeting into the endocytic pathway. For both human immunodeficiency virus (HIV) and Kaposi sarcoma-associated herpesvirus (KSHV), the proteins required for this process are identified, Nef and K3/K5 respectively. The extensive studies in this field have shown that the mechanisms by which these proteins « parasite » the endocytic pathway are completely different. Nef directly interacts with components of the cellular machinery involved in the vesicular transport between the endocytic compartments, mainly the clathrin adaptor complexes (AP), inducing the misrouting of numerous cellular proteins, including CD4, MHC-I, LIGHT, DC-SIGN, CD28 and MHC-II to the endosomal degradation compartment or the trans Golgi-network. The K3 and K5 proteins from KSHV act by inducing the ubiquitylation of the target proteins, such as CMH-I and B7.2, triggering their internalization and subsequent degradation by the highly conserved Tsg101/vps23 ubiquitin-dependent endosomal pathway.
While these findings show that the strategies used by viruses to target cellular proteins to the endocytic pathway are extremely diverse, additional investigations are needed for the complete understanding of the specific roles of Nef and K3/K5 in the physiopathology of HIV and KSHV infections, respectively. In addition, these viral factors represent valuable tools to study the pathway they are perturbing.
Article body
Les pathogènes intracellulaires ont développé des outils moléculaires d’une étonnante efficacité pour détourner les ressources cellulaires à leur profit et leur permettre de se multiplier tout en échappant à la surveillance du système immunitaire. L’un des mécanismes d’échappement qui a été le plus étudié et le mieux caractérisé est la modulation de l’expression de surface des molécules d’histocompatibilité de classe I (CMH-I) dans des modèles d’infections virales. Ces études ont démontré la diversité des mécanismes mis en oeuvre aboutissant à une perturbation de la voie de biosynthèse ou de la voie de transport vers la membrane plasmique [1]. Dans cet article, nous nous sommes intéressés, en détaillant deux exemples, aux virus qui ont plus spécifiquement ciblé la voie d’endocytose : (1) le virus de l’immunodéficience humaine (VIH) qui, par sa protéine Nef (Nef pour negative factor), interagit avec plusieurs éléments de la machinerie de la voie d’endocytose pour moduler l’expression de son récepteur CD4, des molécules du CMH-I, mais également d’autres protéines membranaires ; (2) le virus de l’Herpès associé au sarcome de Kaposi (KSHV ou HHV8), dont les protéines K3 et K5 induisent l’internalisation du CMH-I et d’autres molécules d’intérêt immunologique en provoquant leur ubiquitinylation.
VIH : Nef, un perturbateur général de la voie d’endocytose
Accessoire à la réplication virale in vitro, la protéine Nef des VIH joue un rôle essentiel in vivo dans la physiopathologie de l’infection et l’induction du syndrome d’immunodéficience acquise (SIDA) [2]. Comme les autres protéines accessoires des VIH, Nef est une protéine pluri-fonctionnelle qui intervient au cours de l’infection en perturbant des voies métaboliques essentielles au fonctionnement de la cellule infectée afin d’optimiser la réplication virale et de limiter les mécanismes de défense de l’hôte.
L’activité de Nef la plus documentée est sa capacité de moduler l’expression d’un certain nombre de protéines à la surface des cellules infectées. Révélée initialement par l’induction d’une diminution importante de l’expression membranaire de CD4, puis des molécules du CMH-I, cette activité concerne également d’autres protéines membranaires ; les molécules de classe II du CMH (CMH-II), la molécule co-stimulatrice CD28, les cytokines membranaires TNF (tumor necrosis factor) et LIGHT et la lectine DC-SIGN (dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin) exprimée à la surface des cellules dendritiques [3-5]. L’effet de Nef se traduit par une diminution de l’expression membranaire des molécules CD28 et du CMH-II matures et, inversement, par une augmentation de l’expression de TNF, LIGHT et DC-SIGN, ainsi que des molécules du CMH-II immatures. Ces effets de Nef décrits dans des modèles in vitro pourraient rendre compte de son importance comme facteur de virulence in vivo [2]. La réduction de l’expression de surface de molécules du CMH-I (HLA-A et HLA-B) et CMH-II fonctionnelles pourrait constituer un mécanisme d’échappement à la réponse immunitaire, alors que l’absence d’effet sur les molécules HLA-C protégerait les cellules infectées de la lyse par les cellules NK (natural killer). La modulation de l’expression de CD4 contribue à l’augmentation de l’infectivité virale en favorisant le bourgeonnement des virions à la surface cellulaire, mais également en permettant une incorporation optimale des glycoprotéines d’enveloppe dans les particules virales [2]. Enfin, l’augmentation de l’expression de DC-SIGN à la surface des cellules dendritiques augmenterait l’adhérence des lymphocytes T afin de faciliter la transmission virale [5].
Figure 1
Nef : protéines cibles, structure et interactions avec la voie d’endocytose.
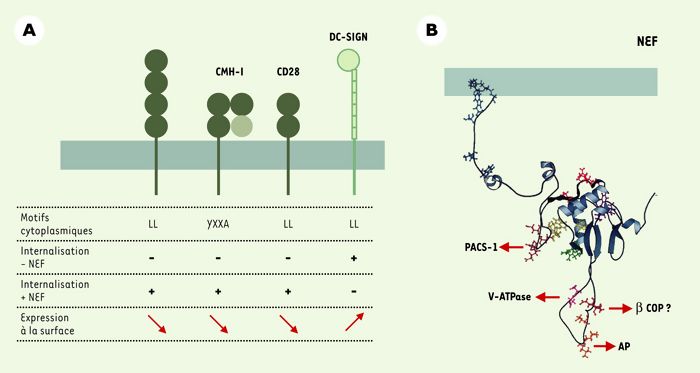
A. Protéines membranaires dont l’expression de surface est modulée par Nef. Les motifs cytoplasmiques des protéines cibles et le rôle de ces motifs en présence ou en l’absence de Nef sont indiqués. Nef diminue l’expression de surface des molécules CD4, CMH-I et CD28 alors qu’elle augmente l’expression de DC-SIGN. CD4 : co-récepteur du VIH ; CD28 : molécule co-activatrice, ligand de B7 ; CMH-I : molécules du complexe majeur d’histocompatibilité de classe I ; DC-SIGN : dendritic cell-specific intercellular adhesion molecule 3- grabbing nonintegrin. B. Nef interagit avec différentes protéines de la machinerie de la voie d’endocytose. Les motifs impliqués dans ces interactions sont indiqués sur la structure tridimensionnelle de Nef VIH-1. AP : clathrin assembly protein complexes ; β-COP : sous-unité β du complexe COPI ; PACS-1 : phosphofurine acidic cluster sorting protein 1 ; v-ATPase : sous-unité catalytique de l’ATPase vacuolaire (d’après [17]).
À la lecture des résultats récents, les effets de Nef ne peuvent plus être expliqués simplement par une augmentation spécifique de l’internalisation de certains récepteurs membranaires puisque des effets opposés sont observés en fonction du récepteur étudié. Les molécules CD4, CD28 et CMH-I sont exprimées de façon stable à la membrane et Nef induit leur internalisation, alors qu’elle stabilise DC-SIGN qui est normalement internalisée de façon constitutive. Les mécanismes responsables de cette différence ne sont a priori pas évidents à expliquer puisqu’ils passent par des motifs comparables (Figure 1A). L’internalisation des molécules HLA-A et HLA-B du CMH-I induite par Nef est liée à la présence dans leur domaine cytoplasmique d’un résidu tyrosine au sein d’un motif YXXA (X représente un résidu quelconque) [6], celle de CD4 et de CD28, à un motif de type di-leucine (di-Leu) [7, 8]. Ces deux types de motifs ressemblent aux signaux de tri présents dans le domaine cytoplasmique de nombreuses protéines membranaires et responsables de leur trafic au sein de la voie d’endocytose. Ces signaux sont normalement reconnus par les complexes adaptateurs (AP), responsables du trafic vésiculaire entre le réseau trans-golgien (TGN) et les endosomes, de l’internalisation à partir de la membrane plasmique, et du transport vers les endosomes tardifs/lysosomes (Figure 2). Cependant, les motifs indispensables aux effets de Nef ne sont normalement pas fonctionnels, et Nef permettrait leur reconnaissance par la machinerie d’endocytose. Un motif di-Leu de la région cytoplasmique de DC-SIGN est également responsable de l’augmentation de son expression de surface. Contrairement à ceux de CD4 et CD28, ce motif di-Leu est un signal d’internalisation fonctionnel dont la fonction est inhibée par l’expression de Nef expliquant la stabilisation de DC-SIGN à la membrane [5].
Figure 2
Nef VIH-1 et K3/K5 KSHV et voie d’endocytose.

Les rôles respectifs des complexes AP et COPI dans le transport vésiculaire entre les différents compartiments de la voie d’endocytose sont indiqués par des flèches en respectant les codes couleurs utilisés pour la structure. Les voies induites par Nef et par les protéines K3/K5 sont indiquées par des flèches en pointillés noir et mauve, respectivement. La structure des complexes AP et COPI est schématisée au bas de la figure. AP : clathrin assembly protein complexes ; β-COP : sous-unité β du complexe COPI ; CD4 : co-récepteur du VIH ; CMH-I : molécules du complexe majeur d’histocompatibilité de classe I ; COP : coatomer protein complex ; K3/K5 : protéines K3 et K5 du virus associé au sarcome de Kaposi ; Nef : negative factor ; TGN : trans-Golgi network ; Ub : ubiquitinylation.
Les travaux réalisés au cours de ces dernières années ont permis des avancées significatives pour la compréhension des mécanismes responsables des perturbations du trafic intracellulaire induites par Nef. En effet, Nef interagit directement avec les sous-unités μ et β des complexes AP-1, AP-2 et AP-3 [6, 9, 10], ainsi qu’avec la sous-unité β des complexes COPI (coatomer protein complex I) [11, 12] impliqués dans le trafic vésiculaire au sein de la voie de sécrétion, mais qui interviennent également dans le transport entre endosomes précoces et tardifs [13]. Des interactions de Nef avec la sous-unité catalytique de l’ATPase vacuolaire (V-ATPase) nécessaire à l’acidification des compartiments endosomiques, ainsi qu’avec la protéine PACS-1 ont également été rapportées [14, 15]. PACS-1 interagit avec les complexes AP-1 et AP-3 et participe au trafic entre endosomes et TGN [16]. Les déterminants de Nef impliqués dans ces interactions ont été bien caractérisés (Figure 1B). À l’exception d’un groupe de résidus glutamiques localisé dans son tiers N-terminal nécessaire pour l’association à PACS-1 [14], les éléments impliqués dans le recrutement des complexes AP, COPI et de la v-ATPase se trouvent distribués dans sa partie C-terminale au sein d’une région contenant une structure en boucle. Les interactions avec les chaînes μ1 et μ3 des complexes AP-1 et AP-3 s’effectuent par deux régions localisées de part et d’autre de la boucle, alors qu’un motif di-Leu présent dans la partie centrale de la boucle serait responsable de l’association aux sous-unités β1 et β3 [2, 17]. La boucle flexible de Nef est également impliquée, par deux motifs di-acide distincts, dans les interactions respectives avec la V-ATPase et β-COP, même si des résultats contradictoires ont été rapportés pour l’association à β-COP [12, 18].
Dans le cas de la modulation de CD4, un modèle a été proposé dans lequel Nef utilise séquentiellement les différentes machineries de la voie d’endocytose, et joue donc le rôle de connecteur entre les molécules CD4 et ces différentes machineries [19]. La modulation de l’expression de surface de CD4 résulterait d’un processus en deux étapes (Figure 2). À la membrane plasmique, Nef recrute les molécules CD4 dans les puits recouverts de clathrine par son interaction avec AP-2, induisant ainsi son internalisation et son adressage vers les endosomes précoces. L’association de Nef aux complexes COP-I permet ensuite le ciblage de CD4 vers les compartiments de dégradation (endosomes tardifs/lysosomes). Ce modèle pourrait également expliquer les effets de Nef sur l’internalisation de CD28 [8]. Pour les molécules du CMH-I, le même type de modèle séquentiel a été proposé avec toutefois des différences importantes sur les mécanismes mis en oeuvre par Nef. En effet, l’internalisation des molécules du CMH-I ne semble pas passer par la voie dépendante des puits recouverts de clathrine [20]. La seconde différence avec le modèle CD4 est que les molécules de classe I ne sont pas dirigées vers les compartiments de dégradation mais retournent vers le TGN, par la connexion de Nef à la protéine PACS-1 [14] (Figure 2).
Ces modèles permettent d’expliquer les effets généraux de Nef sur ses différentes protéines cibles, mais de nombreuses interrogations restent posées sur les mécanismes responsables de chacune des étapes. Les motifs di-Leu de CD4 et CD28 et les motifs YXXA des molécules HLA-A et HLA-B sont indispensables aux effets de Nef. Ces motifs ne sont normalement pas reconnus par la machinerie d’endocytose, Nef pourrait alors interagir directement avec ces signaux et les connecter aux différentes protéines de cette machinerie. Toutefois, les données concernant des interactions directes de Nef avec les signaux de CD4, CD28 et CMH-I sont extrêmement minces ou inexistantes [19]. Alternativement, Nef pourrait modifier les propriétés de reconnaissance de ces signaux par les protéines de la machinerie d’endocytose. Cependant, des données récentes montrent qu’un motif di-Leu est également responsable des effets de Nef sur l’inhibition de l’internalisation de DC-SIGN [5], suggérant que Nef perturbe dans ce cas la reconnaissance du motif. D’autres limitations de ces modèles peuvent être discutées. La première étape implique en effet une interaction avec AP-2, alors que la protéine Nef des VIH-1 recrute préférentiellement AP-1 et AP-3 [9, 10, 21]. Ces résultats semblent en contradiction avec les effets de Nef principalement centrés sur l’internalisation à la membrane plasmique, et pourrait suggérer un re-routage direct du TGN vers les endosomes (Figure 2). Le rôle des complexes COPI dans la dégradation de CD4 est également remis en question en raison des résultats contradictoires sur le rôle du motif di-acide de Nef dans l’association à β-COP [12, 18]. Cette seconde étape de ciblage vers les compartiments de dégradation pourrait alternativement être reliée au recrutement des complexes AP-3 qui participent au transport vésiculaire vers ces compartiments. Enfin, le recrutement forcé des manteaux AP-1, AP-3 et COP-I sur les membranes endosomiques pourrait expliquer l’expansion importante de ce compartiment induite par l’expression de Nef [21, 22].
En interagissant avec de nombreux éléments de la machinerie d’endocytose, Nef se comporte comme un perturbateur général de cette voie de transport. Les différents points discutés dans cet article indiquent que les contributions respectives et la dynamique des interactions restent en grande partie à préciser pour comprendre les mécanismes d’action de Nef.
KSHV : internalisation forcée par ubiquitinylation
Le KSHV appartient à la famille des herpesviridae, et est associé au développement du sarcome de Kaposi et d’autres pathologies tumorales rencontrées notamment chez les patients infectés par le VIH [23]. Il était déjà connu que les virus de cette famille échappent au système immunitaire entre autres en modulant l’expression de surface du CMH-I par différents mécanismes, soit très tôt dans la voie de biosynthèse, soit en déroutant les molécules néo-synthétisées vers les lysosomes [1]. Récemment, plusieurs équipes ont démontré que le KSHV présente une activité similaire portée par deux protéines, K3 et K5, suffisantes pour diminuer l’expression de surface du CMH-I et d’autres molécules de la réponse immune, B7.2 (co-stimulation) et ICAM-1 (intercellular adhesion molecule 1). Les mécanismes mis en oeuvre par KSHV sont différents de ceux connus pour les virus apparentés car cette diminution d’expression n’est pas due à une perturbation précoce de la voie de biosynthèse. Les molécules de classe I sont correctement synthétisées et exportées à la membrane plasmique, mais leur internalisation est augmentée [24-27].
Figure 3
K3/K5 : protéines cibles, structure et interactions avec la machinerie d’ubiquitinylation.
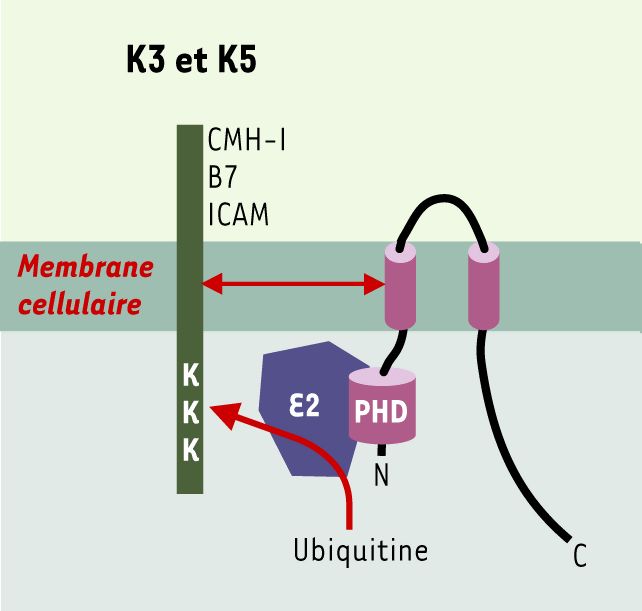
Les protéines K3 et K5 possèdent des structures semblables avec des domaines N- et C-terminaux cytoplasmiques, deux domaines transmembranaires et une courte boucle luminale. Elles présentent dans leur région N-terminale un domaine PHD/LAP qui interagit avec une ubiquitine ligase de type E2 responsable de l’ubiquitinylation des protéines cibles. B7 : molécule co-activatrice, ligand de CD28 ; ICAM : intercellular adhesion molecule ; K3/K5 : protéines K3 et K5 du virus associé au sarcome de Kaposi ; PHD/LAP : plant homeo domain/leukemia associated protein (d’après [28]).
K3 et K5 sont des protéines homologues dont la localisation intracellulaire semble être restreinte au réticulum endoplasmique [24, 25], ne permettant pas de comprendre a priori leurs effets à la membrane. Des travaux récents ont permis de mieux appréhender leur mode d’action, elles induisent une ubiquitinylation des protéines cibles sur des résidus lysines de leur région cytoplasmique, provoquant ainsi leur internalisation et leur dégradation dans le compartiment endosomique tardif [28]. Les données récentes montrant que la dégradation des molécules du CMH-I ubiquitinylées par K3 est dépendante de la protéine tsg-101 (tumor susceptibility gene-101) [29] confirment que le KSHV utilise la voie de dégradation dépendante de l’ubiquitine impliquée dans la dégradation d’un nombre croissant de récepteurs de facteurs de croissance [30, 31]. Ces données démontrent que l’ubiquitinylation de récepteurs normalement stables à la membrane est suffisante pour déclencher leur internalisation.
L’ubiquitinylation des protéines cibles dépend d’un domaine de type PHD/LAP présent dans la région cytoplasmique N-terminale qui reconnaît une ubiquitine ligase de type E2 directement responsable des événements d’ubiquitinylation (Figure 3). Les protéines K3 et K5 permettraient la reconnaissance directe des substrats. Ainsi, K3 présente un spectre plus large que K5 sur l’ensemble des molécules du CMH-I, K5 ne module que HLA-A et HLA-B. À l’opposé, seule l’expression de K5 module l’expression de B7.2 et d’ICAM-1. Ces différences de spécificité semblent passer par une interaction entre les régions trans-membranaires de K3 et K5 et celles des protéines cibles [26-28].
Le virus murin homologue de KSHV, MHV-68, module aussi l’expression de surface du CMH-I par l’intermédiaire de protéines semblables à K3 et K5 (MK3 et MK5) responsables de l’ubiquitinylation des molécules de classe I murines. De façon surprenante, l’effet de cette même modification conduit dans le système murin à une dégradation précoce des molécules néo-synthétisées par le protéasome avant qu’elles ne quittent le réticulum endoplasmique [32]. Une telle différence dans des systèmes si proches conduit à se poser la question de l’intérêt pour les virus de cibler l’une ou l’autre de ces voies pour aboutir à un résultat apparemment identique. À l’opposé, on peut imaginer que la nature des voies empruntées n’est pas importante mais que c’est le résultat, c’est-à-dire la modulation de certaines molécules cibles, qui compte.
Les exemples de virus qui ont développé des systèmes d’induction d’internalisation de protéines membranaires sont plutôt rares. L’adénovirus humain constitue cependant un autre exemple de modulation de l’expression de récepteurs impliqués dans l’apoptose tels que Fas ou le récepteur de TRAIL [33, 34]. Cette modulation passe aussi par l’expression de deux protéines virales appelées RID (receptor internalization and degradation) α et β (ou protéines E3 10.4K et 14.5K) qui avaient été initialement décrites pour induire l’internalisation du récepteur de l’EGF [35]. Ces protéines induisent l’internalisation de Fas et de TRAIL-R et leur dégradation dans les compartiments acides. Les mécanismes impliqués ne sont pas connus. Il est toutefois intéressant de noter que des similitudes existent entre ces deux systèmes, puisque RID α et β sont également localisées au niveau du réticulum endoplasmique. De plus, l’expression des protéines RID à la membrane plasmique n’est observée que lorsque α et β sont co-exprimées [34], ce qui pourrait être aussi le cas pour K3 et K5.
Conclusions
Il semble que les exemples de virus utilisant la voie d’endocytose pour moduler l’expression de molécules de surface sont tout aussi divers que ceux ayant « choisi » de dérouter leurs protéines cibles très tôt au cours de leur biosynthèse. Les deux exemples décrits ici sont intéressants car alors que ces deux virus sont capables de moduler l’internalisation de protéines de surface, ils ont « trouvé » des solutions différentes pour arriver à leurs fins. Le VIH interagit avec la machinerie cellulaire responsable du transport vésiculaire au sein de la voie d’endocytose alors que KSHV modifie certaines protéines cibles les rendant apte à emprunter cette voie. Ce dernier système semble plus spécifique a priori car seules les protéines pouvant être ubiquitinylées par K3 et K5 seront internalisées et dégradées. Dans le cas de Nef, on peut, en revanche, s’attendre à des effets collatéraux plus importants résultant de la perturbation des fonctions exercées par les manteaux vésiculaires avec lesquels elle interagit et donc à des altérations plus générales du trafic des protéines au sein de la voie d’endocytose.
Appendices
Remerciements
Les travaux réalisés dans les laboratoires des auteurs ont été partiellement subventionnés par la Fondation pour la Recherche Médicale et le contrat ARC 5807 (A.B.) et par des contrats Sidaction et ANRS (S.B.).
Références
- 1. Tortorella D, Gewurz BE, Furman MH, et al. Viral subversion of the immune system. Annu Rev Immunol 2000 ; 18 : 861-926.
- 2. Janvier K, Petit C, Le Rouzic E, et al. HIV auxiliary proteins : an interface between the virus and the host. AIDS 2000 ; 14 : S21-30.
- 3. Lama J, Ware CF. Human immunodeficiency virus type 1 Nef mediates sustained membrane expression of tumor necrosis factor and the related cytokine Light on activated T cells. J Virol 2000 ; 74 : 9396-402.
- 4. Stumptner-Cuvelette P, Morchoisne S, Dugast M, et al. HIV-1 Nef impairs MHC class II antigen presentation and surface expression. Proc Natl Acad Sci USA 2001 ; 98 : 12144-9.
- 5. Sol-Foulon N, Moris A, Nobile C, et al. HIV-1 Nef-induced upregulation of DC-Sign in dendritic cells promotes lymphocyte clustering and viral spread. Immunity 2002 ; 16 : 145-55.
- 6. Le Gall S, Erdtmann L, Benichou S, et al. Nef interacts with the mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity 1998 ; 8 : 483-95.
- 7. Aiken C, Konner J, Landau NR, et al. Nef induces CD4 endocytosis : requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell 1994 ; 76 : 853-64.
- 8. Swigut T, Shohdy N, Skowronski J. Mechanism for down-regulation of CD28 by Nef. EMBO J 2001 ; 20 : 1593-604.
- 9. Bresnahan PA, Yonemoto W, Ferrell S, et al. A dileucine motif in HIV-1 Nef acts as an internalization signal for CD4 downregulation and binds the AP-1 clathrin adaptor. Curr Biol 1998 ; 8 : 1235-8.
- 10. Craig HM, Reddy TR, Riggs NL, et al. Interactions of HIV-1 nef with the mu subunits of adaptor protein complexes 1, 2, and 3 : role of the dileucine-based sorting motif. Virology 2000 ; 271 : 9-17.
- 11. Benichou S, Bomsel M, Bodeus M, et al. Physical interaction of the HIV-1 Nef protein with beta-COP, a component of non-clathrin-coated vesicles essential for membrane traffic. J Biol Chem 1994 ; 269 : 30073-6.
- 12. Piguet V, Gu F, Foti M, et al. Nef-induced CD4 degradation : a diacidic-based motif in Nef functions as a lysosomal targeting signal through the binding of beta-COP in endosomes. Cell 1999 ; 97 : 63-73.
- 13. Aniento F, Gu F, Parton RG, Gruenberg J. An endosomal beta COP is involved in the pH-dependent formation of transport vesicles destined for late endosomes. J Cell Biol 1996 ; 133 : 29-41
- 14. Piguet V, Wan L, Borel C, et al. HIV-1 Nef protein binds to the cellular protein PACS-1 to downregulate class I major histocompatibility complexes. Nat Cell Biol 2000 ; 2 : 163-7.
- 15. Lu X, Yu H, Liu SH, et al. Interactions between HIV-1, Nef and vacuolar ATPase facilitate the internalization of CD4. Immunity 1998 ; 8 : 647-56.
- 16. Crump CM, Xiang Y, Thomas L, et al. PACS-1 binding to adaptors is required for acidic cluster motif-mediated protein traffic. EMBO J 2001 ; 20 : 2191-201.
- 17. Geyer M, Fackler OT, Peterlin BM. Structure-function relationships in HIV-1 Nef. EMBO Rep 2001 ; 2 : 580-5.
- 18. Janvier K, Craig H, Le Gall S, et al. Nef-induced CD4 downregulation : a diacidic sequence in human immunodeficiency virus type 1 Nef does not function as a protein sorting motif through direct binding to beta-COP. J Virol 2001 ; 75 : 3971-6.
- 19. Doms RW, Trono D. The plasma membrane as a combat zone in the HIV battlefield. Genes Dev 2000 ; 14 : 2677-88.
- 20. Le Gall S, Buseyne F, Trocha A, et al. Distinct trafficking pathways mediate Nef-induced and clathrin-dependent major histocompatibility complex class I down-regulation. J Virol 2000 ; 74 : 9256-66.
- 21. Erdtmann L, Janvier K, Raposo G, et al. Two independent regions of HIV-1 Nef are required for connection with the endocytic pathway through binding to the mu 1 chain of AP1 complex. Traffic 2000 ; 1 : 871-83.
- 22. Sanfridson A, Hester S, Doyle C. Nef proteins encoded by human and simian immunodeficiency viruses induce the accumulation of endosomes and lysosomes in human T cells. Proc Natl Acad Sci USA 1997 ; 94 : 873-8.
- 23. Choi J, Means RE, Damania B, et al. Molecular piracy of Kaposi’s sarcoma associated herpesvirus. Cytokine Growth Factor Rev 2001 ; 12 : 245-57.
- 24. Coscoy L, Ganem D. Kaposi’s sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc Natl Acad Sci USA 2000 ; 97 : 8051-6.
- 25. Ishido S, Wang C, Lee BS, et al. Downregulation of major histocompatibility complex class I molecules by Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins. J Virol 2000 ; 74 : 5300-9.
- 26. Coscoy L, Ganem D. A viral protein that selectively downregulates ICAM-1 and B7-2 and modulates T cell costimulation. J Clin Invest 2001 ; 107 : 1599-606.
- 27. Ishido S, Choi JK, Lee BS, et al. Inhibition of natural killer cell-mediated cytotoxicity by Kaposi’s sarcoma-associated herpesvirus K5 protein. Immunity 2000 ; 13 : 365-74.
- 28. Coscoy L, Sanchez DJ, Ganem D. A novel class of herpesvirus-encoded membrane-bound E3 ubiquitin ligases regulates endocytosis of proteins involved in immune recognition. J Cell Biol 2001 ; 155 : 1265-73.
- 29. Hewitt EW, Duncan L, Mufti D, Baker J, Stevenson PG, Lehner PJ. Ubiquitylation of MHC class I by the K3 viral protein signals internalization and TSG101-dependent degradation. EMBO J 2002 ; 21 : 2418-29.
- 30. Hicke L. Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol 2001 ; 2 : 195-201.
- 31. Conibear E. An ESCRT into the endosome. Mol Cell 2002 ; 10 : 215-6.
- 32. Boname JM, Stevenson PG. MHC class I ubiquitination by a viral PHD/LAP finger protein. Immunity 2001 ; 15 : 627-36.
- 33. Tollefson AE, Hermiston TW, Lichtenstein DL, et al. Forced degradation of Fas inhibits apoptosis in adenovirus-infected cells. Nature 1998 ; 392 : 726-30.
- 34. Tollefson AE, Toth K, Doronin K, et al. Inhibition of Trail-induced apoptosis and forced internalization of Trail receptor 1 by adenovirus proteins. J Virol 2001 ; 75 : 8875-87.
- 35. Carlin CR, Tollefson AE, Brady HA, et al. Epidermal growth factor receptor is down-regulated by a 10,400 MW protein encoded by the E3 region of adenovirus. Cell 1989 ; 57 : 135-44.
List of figures
Figure 1
Nef : protéines cibles, structure et interactions avec la voie d’endocytose.
A. Protéines membranaires dont l’expression de surface est modulée par Nef. Les motifs cytoplasmiques des protéines cibles et le rôle de ces motifs en présence ou en l’absence de Nef sont indiqués. Nef diminue l’expression de surface des molécules CD4, CMH-I et CD28 alors qu’elle augmente l’expression de DC-SIGN. CD4 : co-récepteur du VIH ; CD28 : molécule co-activatrice, ligand de B7 ; CMH-I : molécules du complexe majeur d’histocompatibilité de classe I ; DC-SIGN : dendritic cell-specific intercellular adhesion molecule 3- grabbing nonintegrin. B. Nef interagit avec différentes protéines de la machinerie de la voie d’endocytose. Les motifs impliqués dans ces interactions sont indiqués sur la structure tridimensionnelle de Nef VIH-1. AP : clathrin assembly protein complexes ; β-COP : sous-unité β du complexe COPI ; PACS-1 : phosphofurine acidic cluster sorting protein 1 ; v-ATPase : sous-unité catalytique de l’ATPase vacuolaire (d’après [17]).
Figure 2
Nef VIH-1 et K3/K5 KSHV et voie d’endocytose.
Les rôles respectifs des complexes AP et COPI dans le transport vésiculaire entre les différents compartiments de la voie d’endocytose sont indiqués par des flèches en respectant les codes couleurs utilisés pour la structure. Les voies induites par Nef et par les protéines K3/K5 sont indiquées par des flèches en pointillés noir et mauve, respectivement. La structure des complexes AP et COPI est schématisée au bas de la figure. AP : clathrin assembly protein complexes ; β-COP : sous-unité β du complexe COPI ; CD4 : co-récepteur du VIH ; CMH-I : molécules du complexe majeur d’histocompatibilité de classe I ; COP : coatomer protein complex ; K3/K5 : protéines K3 et K5 du virus associé au sarcome de Kaposi ; Nef : negative factor ; TGN : trans-Golgi network ; Ub : ubiquitinylation.
Figure 3
K3/K5 : protéines cibles, structure et interactions avec la machinerie d’ubiquitinylation.
Les protéines K3 et K5 possèdent des structures semblables avec des domaines N- et C-terminaux cytoplasmiques, deux domaines transmembranaires et une courte boucle luminale. Elles présentent dans leur région N-terminale un domaine PHD/LAP qui interagit avec une ubiquitine ligase de type E2 responsable de l’ubiquitinylation des protéines cibles. B7 : molécule co-activatrice, ligand de CD28 ; ICAM : intercellular adhesion molecule ; K3/K5 : protéines K3 et K5 du virus associé au sarcome de Kaposi ; PHD/LAP : plant homeo domain/leukemia associated protein (d’après [28]).